Список первичных иммунодефицитов
Это список первичных иммунодефицитов (ПИД), которые представляют собой иммунодефициты , которые не являются вторичными по отношению к другому состоянию.
Международный союз иммунологических обществ признает девять классов первичных иммунодефицитов, всего около 430 состояний. [ 1 ] [ 2 ] В обновлении руководства по классификации 2014 года добавлена 9-я категория и добавлено 30 новых генных дефектов из предыдущей версии 2009 года. [ 3 ] [ 4 ] Последняя классификация была выпущена в 2019 году. [ 5 ] Число выявленных состояний продолжает расти с течением времени по мере проведения дополнительных исследований.
Последствия первичных иммунодефицитов варьируются от легких до тяжелых в зависимости от состояния. [ 6 ]
Комбинированный Т- и В-клеточный иммунодефицит
[ редактировать ]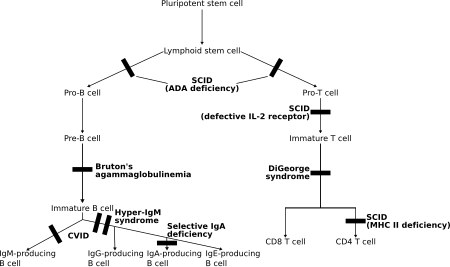
При этих заболеваниях как Т-лимфоциты , так и часто В-лимфоциты , регуляторы адаптивного иммунитета, дисфункциональны или уменьшены в количестве. Основными представителями являются различные типы тяжелого комбинированного иммунодефицита (ТКИД). [ 7 ]
- Т-/В+ ТКИД (преимущественно отсутствуют Т-клетки):
- дефицит γc
- Дефицит JAK3
- рецептора интерлейкина-7-α Дефицит
- CD45 Дефицит
- CD3δ, CD3ε или CD3ζ Дефицит
- коронина-1А Дефицит
- LAT (гена) Дефицит
- Т-/В-SCID (отсутствуют как Т-, так и В-клетки)
- RAG 1/2 Дефицит
- Дефицит DCLRE1C (Артемида)
- XLF (белок) / дефицит Цернунноса
- ДНК PKcs Дефицит
- ДНК-лигазы IV типа Дефицит
- дефицит аденозиндезаминазы (АДА)
- ретикулярная дисгенезия
- Синдром Омена
- лиганда CD40 Дефицит
- CD40 Дефицит
- CD3γ дефицит
- CD8 Дефицит
- ICOS Дефицит
- ZAP70 Дефицит
- Дефицит каналов Ca++
- Дефицит MHC класса I (с мутациями в TAP1 , TAP2 , TAPBP или B2M )
- Дефицит MHC класса II (с мутациями в CIITA , RFXANK , RFX5 или RFXAP )
- Дефицит CD25
- Дефицит CD27
- Дефицит STAT5b
- Дефицит ИТК
- Дефицит SH2D1A (XLP1)
- Дефицит MAGT1
- DOCK2 Дефицит
- Дефицит DOCK8
- Дефицит RhoH
- Активированный дельта-синдром PI3K
- MALT1 Дефицит
- BCL10 Дефицит
- BCL11B Дефицит
- CARD11 Дефицит
- Дефицит MST1
- Дефицит TCRα
- Дефицит LCK
- IL-21 Дефицит
- Дефицит IL-21R
- Дефицит UNC119
- Дефицит НИК
- Дефицит OX40
- Дефицит ИКБКБ
- TFRC Дефицит
- моезина Дефицит
- RELB Дефицит
- Гипоплазия хрящевых волос
- Дефицит LRBA
Преимущественно дефицит антител
[ редактировать ]При первичном дефиците антител один или несколько изотипов иммуноглобулина уменьшаются или не функционируют должным образом. Эти белки, вырабатываемые плазматическими клетками , обычно связываются с патогенами, направляя их на уничтожение. [ 7 ]
- Отсутствие В-клеток с последующим тяжелым снижением всех типов антител: Х-сцепленная агаммаглобулинемия ( дефицит btk или агаммаглобулинемия Брутона ), дефицит μ - тяжелой цепи , дефицит l 5 , дефицит Igα , BLNK дефицит , тимома с иммунодефицитом.
- В-клетки низкие, но присутствуют или нормальные, но с уменьшением 2 или более изотипов (обычно IgG и IgA, иногда IgM): общий вариабельный иммунодефицит (CVID), дефицит CD19 , дефицит TACI (TNFRSF13B), дефицит рецептора BAFF .
- Нормальное количество В-клеток со снижением IgG и IgA и повышением IgM : синдромы гипер-IgM.
- Нормальное количество B-клеток с дефицитом изотипа или легкой цепи: делеции тяжелых цепей , дефицит каппа-цепи , изолированный дефицит подкласса IgG, IgA с дефицитом подкласса IgG, селективный дефицит иммуноглобулина А.
- Дефицит специфических антител к специфическим антигенам при нормальных В-клетках и нормальной концентрации Ig.
- Транзиторная гипогаммаглобулинемия младенчества (ТГИ)
Другой четко выраженный синдром иммунодефицита
[ редактировать ]Ряд синдромов не поддается формальной классификации, но в остальном их можно распознать по определенным клиническим или иммунологическим признакам. [ 7 ]
- Иммунодефицит с тромбоцитопенией
- Дефекты репарации ДНК , не вызывающие изолированного ТКИД:
- Атаксия-телеангиэктазия
- Атаксия-подобный синдром
- Синдром поломки Неймегена
- Синдром Блума
- Иммунодефицит-центромерная нестабильность-синдром аномалий лица (МКФ1, 2, 3 и 4)
- Дефицит PMS2
- Синдром RIDDLE (дефицит RNF168)
- Дефицит MCM4
- Синдром FILS ( POLE ) дефицит
- POLE2 Дефицит
- LIG1 Дефицит
- NSMCE3 Дефицит
- Дефицит гебо
- GINS1 Дефицит
- Синдром ДиДжорджа (при наличии тимуса ) дефектов
- TBX1 Дефицит
- Синдром CHARGE ( CHD7 дефицит или дефицит SEMA3E )
- Крылатая спираль / FOXN1 дефицит
- Делеция хромосомы 10p13-p14
- Иммуно-костные дисплазии ( аномальное развитие скелета при иммунных проблемах):
- Хрящево-волосовая гипоплазия
- синдром Шимке
- MYSM1 Дефицит
- MOPD1 Дефицит
- EXTL3 Дефицит
- Синдромы гипер IgE
- Синдром Джоба ( STAT3 ) дефицит
- Синдром Кьюта-Нетертона
- PGM3 Дефицит
- Гипогидротическая эктодермальная дисплазия
- Дефекты кальциевых каналов
- ORAI1 Дефицит
- Дефицит STIM1
- транскобаламина 2 Дефицит
- Иммунодефицит с множественными атрезиями кишечника ( дефицит ТТС7А )
- Печеночная веноокклюзионная болезнь с иммунодефицитом (VODI)
- синдром Вичи
- Дефицит пуриннуклеозидфосфорилазы (ПНП)
- АР-ДКС (врожденный аутосомно-доминантный дискератоз)
- Синдром Германского-Пудлака 2 типа.
- Хронический кожно-слизистый кандидоз
- HOIL1 Дефицит
- HOIP Дефицит
- Врожденный XL-дискератоз ( синдром Хойераала-Хрейдарссона )
- Синдром лимфангиэктазии-лимфедемы Хеннека
- Синдром Кабуки
- Дефицит MTHFD1
- Дефицит STAT5b
- Дефицит ИКАРОСа
Болезни иммунной дисрегуляции
[ редактировать ]В определенных условиях преобладающей проблемой является регуляция, а не внутренняя активность частей иммунной системы. [ 7 ]
- Иммунодефицит с гипопигментацией или альбинизмом : синдром Чедиака-Хигаси , синдром Гришелли 2 типа.
- Семейный гемофагоцитарный лимфогистиоцитоз : дефицит перфорина , дефицит UNC13D , синтаксина 11. дефицит
- Х-сцепленный лимфопролиферативный синдром
- Синдромы с аутоиммунитетом:
- (а) Аутоиммунный лимфопролиферативный синдром : тип 1а ( дефекты CD95 ), тип 1b ( дефекты лиганда Fas ), тип 2а ( дефекты CASP10 ), тип 2b ( дефекты CASP8 )
- (б) APECED (аутоиммунная полиэндокринопатия с кандидозом и эктодермальной дистрофией)
- (в) IPEX (иммунодерегуляция, полиэндокринопатия, энтеропатия, Х-сцепленный синдром)
- (г) дефицит CD25
Врожденные дефекты количества фагоцитов, их функции или того и другого.
[ редактировать ]Фагоциты — это клетки, которые поглощают и поглощают патогены ( фагоцитоз ) и уничтожают их с помощью химических веществ. моноциты / макрофаги , так и гранулоциты К этому процессу способны как . В определенных условиях либо снижается количество фагоцитов, либо нарушается их функциональная способность. [ 7 ]
- Тяжелая врожденная нейтропения : из-за дефицита ELA2 (с миелодисплазией )
- Тяжелая врожденная нейтропения : вследствие дефицита GFI1 (с T/B-лимфопенией)
- эластазы Дефицит
- Синдром Костмана ( HAX1 ) дефицит
- Нейтропения с пороками развития сердца и мочеполовой системы.
- Болезнь накопления гликогена, тип 1b.
- синдром Коэна
- Синдром Клерикуцио
- Циклическая нейтропения
- Х-сцепленная нейтропения/миелодисплазия
- Дефицит P14
- HYOU1 Дефицит
- JAGN1 Дефицит
- SMARCD2 Дефицит
- 3-метилглутаконовая ацидурия
- Дефицит адгезии лейкоцитов 1 типа
- Дефицит адгезии лейкоцитов 2 типа
- Дефицит адгезии лейкоцитов 3-го типа
- RAC2 Дефицит ( синдром иммунодефицита нейтрофилов )
- бета-актина Дефицит
- рецептора G-CSF Дефицит
- Локализованный ювенильный пародонтит
- Синдром Папийона-Лефевра
- Специфический дефицит гранул
- Синдром Швахмана-Даймонда
- WDR1 Дефицит
- Муковисцидоз
- Хроническая гранулематозная болезнь : Х-сцепленная или аутосомная ( CYBA , NCF1 , NCF2 , NCF4 )
- IL-12 и IL-23 Дефицит цепи β1
- IL-12p40 Дефицит
- Дефицит глюкозо-6-фосфатдегидрогеназы 1 класса
- рецептора 1 интерферона γ Дефицит
- рецептора интерферона γ 2 Дефицит
- STAT1 Дефицит
- MKL1 Дефицит
- АД гипер-IgE
- АР гипер-IgE
- Легочный альвеолярный протеиноз
- Синдром MonoMac ( дефицит GATA2 )
Дефекты врожденного иммунитета
[ редактировать ]Некоторые редкие состояния возникают из-за дефектов врожденной иммунной системы , которая является основной линией защиты, независимой от более развитых систем, связанных с лимфоцитами. Многие из этих состояний связаны с проблемами кожи. [ 7 ]
- Рецептор интерлейкина 12, дефицит бета 1
- IL-12p40 Дефицит
- рецептора гамма-интерферона 1 Дефицит
- рецептора гамма-интерферона 2 Дефицит
- Дефицит Tyk2
- JAK1 Потеря функции
- ISG15 Дефицит
- RORc Дефицит
- Дефицит STAT1 , мутация усиления функции
- STAT2 Дефицит
- IRF7 Дефицит
- CD16 Дефицит
- IRF8 Дефицит
- IFNAR2 Дефицит
- Недостатки пути TLR
- MDA5 Дефицит
- Верруциформная эпидермодисплазия
- Синдром WHIM (бородавки, гипогаммаглобулинемия, инфекции, миелокатексис)
- EVER1 и EVER2 Дефицит
- Герпетический энцефалит
- CARD9 Дефицит
- Хронический кожно-слизистый кандидоз
- Трипаносомоз
- RPSA Дефицит с врожденной аспленией
- Дефицит HMOX при врожденной асплении
- Дефицит CLCN7 при остеопорозе
- Дефицит OSTM1 при остеопорозе
- Гнойный гидраденит
Аутовоспалительные заболевания
[ редактировать ]Вместо того, чтобы предрасполагать к инфекциям, большинство аутовоспалительных заболеваний приводят к чрезмерному воспалению. Многие из них проявляются в виде синдромов периодической лихорадки . Они могут напрямую поражать различные органы, а также предрасполагать к долговременному повреждению (например, приводя к амилоида ). отложению [ 7 ]
- Семейная средиземноморская лихорадка
- Синдром Айкарди-Гутьера с TREX1 , SAMHD1 или IFIH1 мутациями
- Спондилоэнхондродисплазия с иммунной дисрегуляцией ( мутация ACP5 )
- Васкулопатия, связанная с STING, с началом в младенчестве
- Х-сцепленное сетчатое пигментное расстройство
- USP18 Дефицит
- СВЕЧА (Хронический атипичный нейтрофильный дерматит с липодистрофией)
- Синдром Синглтона-Мертена
- Периодический синдром, связанный с рецептором TNF (TRAPS)
- Синдром гипер-IgD ( дефицит мевалонаткиназы )
- CIAS1 -ассоциированные заболевания:
- NLRP1 Дефицит
- PAPA-синдром ( гнойный стерильный артрит, гангренозная пиодермия, акне)
- ADAM17 Дефицит
- синдром Блау
- Синдром Маджида (хронический рецидивирующий мультифокальный остеомиелит и врожденная дизэритропоэтическая анемия)
- DIRA (дефицит антагониста рецептора IL-1)
- ДИТРА (дефицит антагониста рецептора IL-36)
- CARD14-опосредованный псориаз (CAMPS)
- Херувизм
- Дефект COPA
- Отулипения /ORAS
Дополнить недостатки
[ редактировать ]Система комплемента является частью как врожденной, так и адаптивной иммунной системы; это группа циркулирующих белков, которые могут связывать патогены и образовывать мембраноатакующий комплекс. Дефицит комплемента является результатом отсутствия любого из этих белков. Они могут предрасполагать к инфекциям, а также к аутоиммунным заболеваниям. [ 7 ]
- Дефицит C1q (волчаночноподобный синдром, ревматоидная болезнь, инфекции)
- Дефицит C1r (то же самое)
- Дефицит C1s
- Дефицит C4 (волчаночноподобный синдром)
- Дефицит С2 (волчаночноподобный синдром, васкулит, полимиозит, гноеродные инфекции )
- Дефицит C3 (рецидивирующие гноеродные инфекции )
- Дефицит C5 (нейсериальные инфекции, СКВ)
- Дефицит C6 (то же самое)
- Дефицит C7 (так же, как васкулит)
- Дефицит C8a
- Дефицит C8b
- Дефицит C9 (нейсериальные инфекции)
- Дефицит ингибитора C1 (наследственный ангионевротический отек)
- Дефицит фактора I ( гноеродные инфекции )
- Дефицит фактора Н ( гемолитик-уремический синдром , мембранопролиферативный гломерулонефрит )
- Дефицит фактора D (нейсериальные инфекции)
- Дефицит пропердина (нейсериальные инфекции)
- Дефицит MBP ( гноеродные инфекции )
- Дефицит MASP2
- Дефицит рецептора комплемента 3
- Дефицит белка-кофактора мембраны (CD46)
- Дефицит ингибитора мембраноатакующего комплекса (CD59)
- Пароксизмальная ночная гемоглобинурия
- Дефицит фиколина 3
- Дефицит пропердина
- Дефицит фактора I
- Дефицит фактора H
- Дефицит тромбомодулина
- ЧАПЕЛ болезнь
Фенокопии первичных иммунодефицитов
[ редактировать ]- Аутоиммунный лимфопролиферативный синдром
- Аутоиммунное лейкопролиферативное заболевание, ассоциированное с РАС
- Крупный зернистый лимфоцитоз
- Атипичный гемолитико-уремический синдром
- Хороший синдром
Ссылки
[ редактировать ]- ^ Бусфиха, Азиз; Джидда, Лейла; Пикард, капуцин; Айляль, Фатима; Бобби Гаспар, Х.; Аль-Герц, Валид; Чатила, Талал; Кроу, Яник Дж. (2018). «Фенотипическая классификация первичных иммунодефицитов IUIS 2017» . Журнал клинической иммунологии . 38 (1): 129–143. дои : 10.1007/ s10875-017-0465-8 ISSN 0271-9142 . ПМЦ 5742599 . ПМИД 29226301 .
- ^ Танье, Стюарт Г.; Аль-Герц, Валид; Бусфиха, Азиз; Чатила, Талал; Каннингем-Рандлс, Шарлотта; Эциони, Амос; Франко, Хосе Луис; Холланд, Стивен М.; Кляйн, Кристоф; Морио, Томохиро; Окс, Ханс Д. (01 января 2020 г.). «Врожденные ошибки иммунитета человека: обновленная информация о классификации от экспертного комитета Международного союза иммунологических обществ за 2019 год» . Журнал клинической иммунологии . 40 (1): 24–64. дои : 10.1007/s10875-019-00737-x . ISSN 1573-2592 . ПМЦ 7082301 . ПМИД 31953710 .
- ^ Валид Аль-Герц; Азиз Бусфиха; Жан-Лоран Казанова; и др. (2014). «Первичные иммунодефицитные заболевания: обновленная информация о классификации экспертного комитета по первичным иммунодефицитам Международного союза иммунологических обществ» (PDF) . Границы в иммунологии . 5 (162): 1–33. дои : 10.3389/fimmu.2014.00162 . ПМК 4001072 . ПМИД 24795713 . (Ошибка: дои : 10.3389/fimmu.2014.00460 )
- ^ Нотаранжело Л., Казанова Дж.Л., Конли М.Э. и др. (2006). «Первичные иммунодефицитные заболевания: обновленная информация о заседании Комитета по классификации первичных иммунодефицитных заболеваний Международного союза иммунологических обществ в Будапеште, 2005 г.» . Дж. Аллергическая клиника. Иммунол . 117 (4): 883–96. дои : 10.1016/j.jaci.2005.12.1347 . ПМИД 16680902 .
- ^ Танье, Стюарт Г.; Аль-Герц, Валид; Бусфиха, Азиз; Чатила, Талал; Каннингем-Рандлс, Шарлотта; Эциони, Амос; Франко, Хосе Луис; Холланд, Стивен М.; Кляйн, Кристоф; Морио, Томохиро; Окс, Ханс Д. (01 января 2020 г.). «Врожденные ошибки иммунитета человека: обновленная информация о классификации от экспертного комитета Международного союза иммунологических обществ за 2019 год» . Журнал клинической иммунологии . 40 (1): 24–64. дои : 10.1007/s10875-019-00737-x . ISSN 1573-2592 . ПМЦ 7082301 . ПМИД 31953710 .
- ^ «Общий вариабельный иммунодефицит» . НОРД (Национальная организация по редким заболеваниям) . Проверено 5 марта 2019 г.
- ^ Перейти обратно: а б с д и ж г час Нотаранджело Л.Д., Фишер А., Геха Р.С. и др. (декабрь 2009 г.). «Первичные иммунодефициты: обновление 2009 года: Экспертный комитет Международного союза иммунологических обществ (IUIS) по первичным иммунодефицитам (ПИД)» . Дж. Аллергическая клиника. Иммунол . 124 (6): 1161–78. дои : 10.1016/j.jaci.2009.10.013 . ПМЦ 2797319 . ПМИД 20004777 .