Гликогеное хранение болезней типа I типа I
Эта статья требует дополнительных цитат для проверки . ( февраль 2009 г. ) |
Эта статья написана как исследовательская работа или научный журнал . ( Апрель 2020 г. ) |
| GSD Тип i | |
|---|---|
| Другие имена | Болезнь Герке |
| Символ для хранения гликогена типа I типа I | |
| Произношение | |
| Специальность | Эндокринология , генетика , гематология , иммунология |
| Осложнения | Лакциноцидоз , гиперлипидемия , неалкогольная жировая болезнь печени , гепатоцеллюлярная аденома , воспалительное заболевание кишечника |
| Продолжительность | Продолжительность жизни |
| Типы | Тип IA, тип IB |
| Причины | Аутосомно -рецессивное наследование |
| Диагностический метод | Генетическое тестирование , гипогликемия , гепатомегалия типа IB : нейтропения |
| Уход | Кукурузный крахмал , диета |
| Медикамент | Filgrastim |
| Частота | 1 из 100 000 живорождений |
Гликогеновое хранение болезни I типа I ( GSD I ) - это наследственное заболевание , которое предотвращает печени правильное разрушение крови , которое необходимо для поддержания адекватного уровня сахара в . GSD I разделен на два основных типа: GSD IA и GSD IB, которые различаются по причине, представлению и лечению. Существуют также более редкие подтипы, трансаказы для неорганического фосфата (GSD IC) или глюкозы (GSD ID); Тем не менее, недавнее исследование показывает, что биохимические анализы, используемые для дифференциации GSD IC и GSD ID от GSD IB, не являются надежными и поэтому являются GSD IB. [ 1 ]
GSD IA вызван дефицитом фермента глюкозо -6-фосфатазы ; GSD IB, дефицит в транспортном белке глюкозо-6-фосфатно-транслоказ . Поскольку гликогенолиз является основным метаболическим механизмом, с помощью которого печень снабжает глюкозу в организм во время поста , оба дефицита вызывают тяжелую гипогликемию и со временем избыточное хранение гликогена в печени и (в некоторых случаях) в почках .
Из-за накопления гликогена пациенты с GSD I обычно присутствуют с увеличенной печенью от неалкогольной жировой болезни печени . [ 2 ] Другие функции печени и почки изначально не повреждены в GSD I, но подвержены другим проблемам. [ нечеткий ] Без надлежащего лечения GSD I вызывает хронический низкий уровень сахара в крови, что может привести к чрезмерной молочной кислоте , а также аномально высоким липидам в крови и других проблемах. Частые кормления кукурузного крахмала или других углеводов являются основным лечением всех форм GSD I.
GSD IB также имеет хроническую нейтропения из -за дисфункции в производстве нейтрофилов в костном мозге . Этот иммунодефицит , если не лечить, делает пациентов с GSD IB подверженными инфекции. [ 3 ] Основным лечением этой особенности GSD IB является Filgrastim ; Тем не менее, пациенты часто по -прежнему требуют лечения частых инфекций, а хронически увеличенная селезенка является общим побочным эффектом. [ 4 ] Пациенты с GSD IB часто имеют воспалительное заболевание кишечника . [ 5 ]
Это наиболее распространенный из болезней хранения гликогена . GSD I имеет частоту примерно 1 из 100 000 рождений в американском населении и приблизительно 1 из 20 000 рождений среди евреев ашкенази . [ 6 ] Болезнь была названа в честь немецкого доктора Эдгара фон Гирке , который впервые описал ее в 1929 году. [ 7 ] [ 8 ]
Признаки и симптомы
[ редактировать ]Ранние исследования GSD I выявили многочисленные клинические проявления ложно, как считаются основными признаками генетического заболевания. Тем не менее, продолжающиеся исследования показали, что эти клинические особенности являются последствиями только одного (в GSD IA) или двух (в GSD IB) фундаментальных нарушениях:
- Нарушение способности печени преобразовать сохраненный гликоген в глюкозу с помощью гликогенолиза [ 9 ]
- В GSD IB нарушение способности нейтрофила занимать глюкозу, что приводит к дисфункции нейтрофилов и нейтропении [ 10 ]
Эти фундаментальные нарушения приводят к небольшому числу первичных клинических проявлений, которые являются признаками, рассматриваемыми при диагностике GSD I:
- Низкий уровень сахара в крови (гипогликемия), из -за нарушения гликогенового распада (гликогенолиза), что приводит к недостаточной глюкозе в крови натощак. [ 11 ]
- Гепатомегалия неалкогольной жировой заболевания печени из-за нарушения гликогенолиза, вызывающего накопление гликогена в печени [ 2 ]
- В GSD IB, повышенный риск инфекции из -за нейтропении и дисфункции нейтрофилов [ 3 ]
Пострадавшие люди, обычно присутствующие с вторичными клиническими проявлениями, связанными с одним или несколькими первичными клиническими проявлениями:
- Высокий уровень мочевой кислоты в крови и сопутствующего риска подагры или повреждения почек, вызванный низким уровнем инсулина в сыворотке в длительной гипогликемии
- Высокий уровень молочной кислоты в крови , в экстремальных случаях, приводящих к молочному ацидозу , вызванный длительной гипогликемией [ 11 ]
- печеночные аденомы, развивающиеся во взрослой жизни [ 12 ] и сопровождающий риск анемии , [ 13 ] подозревается, что вызвана дисрегуляцией глюкозы в крови при наличии неалкогольной жировой болезни печени
- В GSD IB, воспалительные заболевания кишечника и сопутствующий риск анемии , [ 13 ] вызвано дисфункцией нейтрофилов и усугубляется увеличенным потреблением углеводов, необходимого для предотвращения гипогликемии [ 3 ]
Кроме того, существует несколько клинических проявлений, которые часто являются результатом лечения первичных клинических проявлений:
- поджелудочной железы Гипертрофия из -за увеличения потребления углеводов, вызывающих частое взаимодействие с ответом инсулина [ 14 ]
- В GSD IB, спленомегалия , из-за долгосрочного использования Filgrastim для лечения нейтропении, вызывая секвестрацию факторов крови в селезенке [ 4 ]
- В GSD IB может возникнуть аномально низкое количество тромбоцитов в крови , из-за долгосрочного использования Filgrastim, вызывая секвестрацию тромбоцитов в селезенке [ 15 ]
- В GSD IB, анемия , из-за долгосрочного использования Filgrastim, вызывающего секвестрацию гемоглобина в селезенке, потенциально усугубленной неконтролируемым воспалительным заболеванием кишечника [ 16 ]
Гипогликемия
[ редактировать ]Низкий уровень сахара в крови (гипогликемия) является основным клиническим симптомом, общим для GSD IA и GSD IB, и чаще всего вызывает первоначальный диагноз заболевания. Во время плода развития при утробном порядке глюкоза матери, переносимая через плаценту, предотвращает гипогликемию . Однако после рождения неспособность поддерживать глюкозу в крови от сохраненного гликогена в печени вызывает измеримую гипогликемию не более чем через 1–2 часа после кормления. Без надлежащего диетического лечения после рождения длительная гипогликемия часто приводит к внезапному лактоцидозу , который может вызвать первичный респираторный дистресс в период новорожденного, а также кетоацидоз . [ Цитация необходима ]
Неврологические проявления гипогликемии менее тяжелы в GSD I, чем в других случаях. Вместо острой гипогликемии, пациенты с GSD I испытывают постоянную легкую гипогликемию. Уменьшенная вероятность неврологических проявлений обусловлена привычкой мозга к легкой гипогликемии. Учитывая снижение уровня глюкозы в крови, мозг адаптируется к использованию альтернативных топлива, такого как лактат . Эти постепенные метаболические адаптации в младенчестве делают тяжелые симптомы, такие как бессознательное или приступ необычайно перед диагнозом. [ Цитация необходима ]
В первые недели жизни недиагностированные младенцы с GSD I переносят постоянную гипогликемию и компенсировал лактоцидоз между кормлением без симптомов. Без постоянного кормления углеводов уровни глюкозы в крови младенческой крови обычно измеряют от 25 до 50 мг/дл (от 1,4 до 2,8 ммоль/л). Через несколько недель до месяцев без лечения последовательными пероральными углеводами младенцы будут прогрессировать, чтобы показать четкие симптомы гипогликемии и лактино -ацидоза. Младенцы могут иметь бледность, хламкость, раздражительность, респираторное расстройство и неспособность спать всю ночь даже на втором курсе жизни. Задержка развития не является внутренним эффектом GSD I, но является общей, если диагноз не понесен в раннем матче. [ Цитация необходима ]
Генетика
[ редактировать ]
GSD I наследуется аутосомно -рецессивным образом. Люди с одной копией неисправного гена являются носителями заболевания и не имеют симптомов. Как и в случае с другими аутосомно -рецессивными заболеваниями, каждый ребенок, рожденный от двух носителей заболевания, имеет 25% вероятность наследования обеих копий неисправного гена и проявить болезнь. Независимые родители ребенка с GSD, я могу предположить, что они являются носителями. Пренатальная диагностика была поставлена биопсией печени плода через 18–22 недели беременности, но лечение плода не было предложено. Пренатальный диагноз возможен с ДНК плода , полученной с помощью хорионной выборки ворсин , когда известно, что плод подвергается риску. [ Цитация необходима ]
Наиболее распространенными формами GSD I являются обозначенные GSD IA и GSD IB, на первом составлении более 80% диагностированных случаев, а второй - менее 20%. Несколько более редких форм были описаны.
- GSD IA является результатом мутаций G6PC , гена для глюкозо-6-фосфатазы, [ 17 ] Расположен на хромосоме 17q 21. [ 18 ]
- GSD IB является результатом мутаций гена для SLC37A4 или «G6PT1», транспортера глюкозы-6-фосфата. [ 18 ] [ 19 ]
- GSD IC является результатом мутаций SLC17A3 или SLC37A4 . [ 20 ]
Глюкозо-6-фосфатаза представляет собой фермент, расположенный на внутренней мембране эндоплазматической ретикулумы . Каталитическая единица связана с кальциевым , связывающим белком кальций , и тремя транспортными белками (T1, T2, T3), которые облегчают движение глюкозо-6-фосфата (G6P), фосфата и глюкозы (соответственно) в и из фермента.
Патофизиология
[ редактировать ]Нормальный баланс углеводов и поддержание уровня глюкозы в крови
[ редактировать ]Гликоген в печени и (в меньшей степени) почки служат формой хранимой, быстро доступной глюкозы, так что уровень глюкозы в крови можно поддерживать между приемами пищи. В течение примерно 3 часов после блюд, содержащего углеводы, высокие уровни инсулина направляют клетки печени, чтобы взять глюкозу из крови, преобразовать ее в глюкозо-6-фосфат (G6P) с ферментом глюкокиназы и добавить молекулы G6P до конца цепочек гликогена (синтез гликогена). Избыточный G6P также отдается в производство триглицеридов и экспортируется для хранения в жировой ткани в качестве жира .
Когда расщепление еды завершено, уровни инсулина падают, а ферментные системы в клетках печени начинают удалять молекулы глюкозы из цепей гликогена в форме G6P. Этот процесс называется гликогенолизом . G6P остается в клетках печени, если фосфат не расщепляется глюкозо-6-фосфатазой. Эта дефосфорилирования реакция дает свободную глюкозу и свободный PO
4 анионы . Свободные молекулы глюкозы могут быть транспортированы из клеток печени в кровь, чтобы поддерживать адекватное снабжение глюкозы в мозг и другие органы организма. Гликогенолиз может обеспечивать потребности взрослого тела в течение 12–18 часов в течение 12–18 часов.
Когда пост продолжается в течение более нескольких часов, падение уровней инсулина допускает белка и катаболизм мышечного триглицеридов из жировой ткани. Продуктами этих процессов являются аминокислоты (в основном аланин ), свободные жирные кислоты и молочная кислота . Свободные жирные кислоты из триглицеридов превращаются в кетоны и в ацетил-КоА . Аминокислоты и молочная кислота используются для синтеза нового G6P в клетках печени в результате процесса глюконеогенеза . Последней стадией нормального глюконеогенеза, как и последняя стадия гликогенолиза, является дефосфорилирование G6P с помощью глюкозо-6-фосфатазы со свободной глюкозой и PO
4 .
Таким образом, глюкозо-6-фосфатаза опосредует конечный, ключевой, шаг в обоих двух основных процессах выработки глюкозы во время поста. Эффект усиливается, потому что результирующие высокие уровни глюкозо-6-фосфата ингибируют более ранние ключевые шаги как в гликогенолизе, так и в глюконеогенезе.
Патофизиология
[ редактировать ]Основными метаболическими эффектами дефицита глюкозо-6-фосфатазы являются гипогликемия , лактоцидоз , гипертриглицеридемия и гиперурикемия .

Гипогликемия GSD I называется «голоданием» или « пост -абсорбционным», обычно примерно через 4 часа после полного пищеварения еды. Эта неспособность поддерживать адекватные уровни глюкозы в крови во время поста вытекает из комбинированного нарушения как гликогенолиза, так и глюконеогенеза. Гипогликемия натощак часто является наиболее важной проблемой в GSD I, и, как правило, проблема, которая приводит к диагностике. Хроническая гипогликемия продуцирует вторичную метаболическую адаптацию, включая хронически низкие уровни инсулина и высокий уровень глюкагона и кортизола .
Лакте ацидоз возникает из -за нарушения глюконеогенеза. Молочная кислота генерируется как в печени, так и в мышцах и окисляется NAD + в пировиновую кислоту , а затем превращается через глюконеогенный путь в G6P. Накопление G6P ингибирует превращение лактата в пируват. Уровень молочной кислоты повышается во время поста, когда падает глюкоза. У людей с GSD I это может не полностью упасть до нормального, даже когда нормальный уровень глюкозы восстановлен.
Гипертриглицеридемия, возникающая в результате амплифицированной продукции триглицеридов, является еще одним косвенным эффектом нарушения глюконеогенеза, амплифицированного хронически низким уровнем инсулина. Во время поста нормальное превращение триглицеридов в свободные жирные кислоты, кетоны и, в конечном счете, нарушено. Уровни триглицеридов в GSD я могу достичь несколько раз нормально и служить клиническим индексом «метаболического контроля».
Гиперурикемия является результатом комбинации повышенной генерации и снижения экскреции мочевой кислоты , которая генерируется, когда увеличение количества G6P метаболизируется через пентозофосфатный путь . Это также побочный продукт деградации пурина . Мочевая кислота конкурирует с молочной кислотой и другими органическими кислотами за почечную экскрецию в моче. В GSD I увеличивала доступность G6P для пентозофосфатного пути, повышение скорости катаболизма и уменьшение экскреции мочи из -за высоких уровней молочной кислоты все объединяется для получения уровней мочевой кислоты в несколько раз нормальных. Хотя гиперурикемия в течение многих лет является бессимптомной, постепенно начисляется повреждение почек и суставов.
Повышенный лактат и лакциницидоз
[ редактировать ]Высокий уровень молочной кислоты в крови наблюдается у всех людей с GSD I из -за нарушения глюконеогенеза . Базовые высоты обычно варьируются от 4 до 10 моль/мл, что не вызовет какого -либо клинического воздействия. Однако во время и после эпизода с низким уровнем сахара в крови уровни лактата внезапно будут расти до превышения 15 моль/мл, порог для лакциноцидоза . Симптомы лактического ацидоза включают рвоту и гиперпною , оба из которых могут усугубить гипогликемию в условиях GSD I. В случаях острого лактоцидоза пациенты нуждаются в неотложной помощи для стабилизации кислорода в крови и восстановления глюкозы в крови. Правильная идентификация лактоцидоза у недиагностированных детей представляет собой проблему, поскольку первыми симптомами обычно являются рвота и обезвоживание, которые имитируют детские инфекции, такие как гастроэнтерит или пневмония . Более того, обе эти общие инфекции могут привести к более тяжелой гипогликемии у недиагностированных детей, что затрудняет диагноз основной причины.
Поскольку повышенный лактат сохраняется, мочевая кислота, кетоциды и свободные жирные кислоты дополнительно увеличивают анионный разрыв . У взрослых и детей высокие концентрации лактата вызывают значительный дискомфорт в мышцах. Этот дискомфорт является усиленной формой ощущения сжигания, которую бегун может ощущать в четырехглавой мышце после спринта, что вызвано кратким накоплением молочной кислоты. Надлежащий контроль гипогликемии в GSD I устраняет возможность лактоцидоза.
Повышенный урат и осложнения
[ редактировать ]Высокие уровни мочевой кислоты часто присутствуют в результате повышенной молочной кислоты у пациентов с GSD I. Когда уровни лактата повышаются, молочной кислоты, передаваемая в крови, конкурирует за тот же механизм транспорта по почкам, что и урат, ограничивая скорость, которую урат может быть очищен по почках в мочу. В случае присутствия, повышенное катаболизм пурина является дополнительным фактором, способствующим. Уровни мочевой кислоты от 6 до 12 мг/дл (от 530 до 1060 umol/l) распространены среди пациентов с GSD I, если заболевание не лечится должным образом. У некоторых пораженных людей использование аллопуринола лекарства необходимо для снижения уровня урата в крови. Последствия гиперурикемии среди пациентов с GSD I включают развитие камней почек и накопление кристаллов мочевой кислоты в суставах, что приводит к заболеванию почек и подагры соответственно.
Гиперлипидемия и плазменные эффекты
[ редактировать ]Повышенные триглицериды в GSD I являются результатом низкого сывороточного инсулина у пациентов с частой длительной гипогликемией. Это также может быть вызвано внутриклеточным накоплением глюкозы-6-фосфата с вторичным шунтированием к пирувату , который превращается в ацетил-КоА , который транспортируется в цитозоль , где происходит синтез жирных кислот и холестерина . Триглицериды выше диапазона 3,4 ммоль/л (300 мг/дл) могут вызывать видимую липемию и даже легкую псевдогипонатриемию из -за уменьшенной водной доли плазмы крови . В GSD I холестерин обычно слегка повышен по сравнению с другими липидами .
Гепатомегалия
[ редактировать ]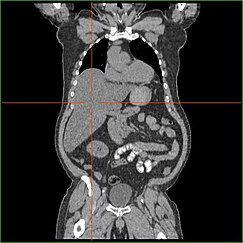
Нарушение способности печени выполнять глюконеогенез приводит к клинически очевидной гепатомегалии . Без этого процесса организм не может освободить гликоген из печени и преобразовать его в глюкозу в крови, что приводит к накоплению хранимого гликогена в печени. Гепатомегалия от накопленного гликогена в печени считается формой неалкогольной жировой болезни печени . Пациенты с GSD I имеют некоторую степень гепатомегалии на протяжении всей жизни, но тяжесть часто связана с потреблением избыточных диетических углеводов . Снижение массы печени возможны, поскольку большинство пациентов сохраняют остаточную функцию печени, которая позволяет освободить сохраненный гликоген с ограниченной скоростью.
Пациенты GSD I часто присутствуют с гепатомегалией со времен рождения. В развитии плода материнская глюкоза, перенесенная на плод, предотвращает гипогликемию, но хранение глюкозы в качестве гликогена в печени приводит к гепатомегалии. Нет никаких доказательств того, что эта гепатомегалия представляет какой -либо риск для правильного развития плода.
Гепатомегалия в GSD -тип I обычно происходит без симпатического увеличения селезенки. Пациенты с GSD IB могут иметь спленомегалию, но это связано с использованием Filgrastim для лечения нейтропении в этом подтипе, а не сопутствующей гепатомегалии. Гепатомегалия будет сохраняться до некоторой степени на протяжении всей жизни, часто вызывая выступление живота, а в тяжелых случаях может быть ощутимо при пупке или ниже . При связанной с GSD неалкогольное жировое заболевание печени обычно функционируют, а ферменты печени и билирубин остаются в нормальном диапазоне. Тем не менее, на функцию печени может повлиять другие осложнения в печени во взрослом возрасте, включая развитие печеночных аденом .
Печеночные аденомы
[ редактировать ]Специфическая этиология печеночных аденом в GSD I остается неизвестной, несмотря на продолжающиеся исследования. Типичный пациент с GSD I, по меньшей мере, с одной аденомой, является взрослым, хотя поражения у пациентов наблюдались в возрасте от четырнадцати лет. Аденомы, состоящие из гетерогенных новообразований, могут происходить индивидуально или в кратных. Оценки о скорости конверсии гепатоцеллюлярной аденомы в гепатоцеллюлярную карциному в GSD I варьируются от 0% до 11%, причем последняя цифра представляет собой более поздние исследования. Одной из причин растущей оценки является растущая популяция пациентов с GSD I, выживающих во взрослую жизнь, когда развивается большинство аденом.
Стандарты лечения диктуют регулярное наблюдение за печенью с помощью МРТ или КТ -сканирования для мониторинга структурных нарушений. Печеночные аденомы могут быть неправильно идентифицированы в качестве очаговой узловой гиперплазии при диагностической визуализации, хотя это состояние встречается редко. Тем не менее, печеночные аденомы в GSD I уникально включают диффузное осаждение гиалинового осаждения маллори , которое в противном случае обычно наблюдается при фокальной узловой гиперплазии. В отличие от общих аденом печени, связанных с пероральной контрацепцией, кровоизлияние у пациентов с GSD I редко.
В то время как причина высокой распространенности аденом в GSD I неясна, исследования с 1970 -х годов приводят сывороточного глюкагона в качестве потенциального водителя . В исследованиях пациенты, которые были помещены в диетический режим, чтобы поддерживать сахар в крови в нормальном диапазоне, охватывающем от 72 до 108 мг/дл (от 4,0 до 6,0 ммоль/л), показали снижение вероятности развития аденомы. Более того, пациенты с хорошо контролируемой глюкозой в крови последовательно наблюдали снижение размера и количества печеночных аденом, что позволяет предположить, что аденомы могут быть вызваны дисбалансом гепатотропных агентов, таких как сывороточный инсулин и особенно сывороточный глюкагон в печени. [ 21 ]
Остеопения
[ редактировать ]У пациентов с GSD у меня часто развивается остеопения . Специфическая этиология низкой минеральной плотности кости в GSD неизвестна, хотя она тесно связана с плохим метаболическим контролем. Остеопения может быть непосредственно вызвана гипогликемией или полученными эндокринными и метаболическими последствиями. Показано, что улучшения в метаболическом контроле постоянно предотвращают или обращают клинически значимую остеопению у пациентов с GSD I. [ 22 ] В тех случаях, когда остеопения прогрессирует с возрастом, плотность минералов кости в ребрах, как правило, более тяжелая, чем в позвонках. [ 23 ] В некоторых случаях Т -показатель минеральной плотности кости будет падать ниже -2,5, что указывает на остеопороз. Есть некоторые доказательства того, что остеопения может быть связана со связанными аномалиями почек в GSD I, особенно гломилярной гиперфильтрации. [ 24 ] Состояние также кажется реагирующим на добавки кальция. Во многих случаях минеральная плотность кости может увеличиваться и возвращаться к нормальному диапазону, учитывая правильный метаболический контроль и только добавки кальция, изменяя остеопению.
Эффекты почек
[ редактировать ]Почки обычно составляют от 10 до 20%, увеличиваемых с сохраненным гликогеном. У взрослых с GSD I хроническое повреждение клубочков , сходное с диабетической нефропатией, может привести к почечной недостаточности . GSD Я могу представить с различными осложнениями почек. Почечные трубчатые аномалии, связанные с гиперлактатемией, наблюдаются в раннем возрасте, вероятно, потому что длительный лактоцидоз, скорее всего, возникает в детстве. Это часто представляется в виде синдрома Фанкони с множественными нарушениями почечной трубчатой реабсорбции, включая трубчатый ацидоз с бикарбонатом и истощением фосфатов. Эти трубчатые нарушения в GSD I обычно обнаруживаются и контролируются кальцием мочи. В долгосрочной перспективе эти расстояния могут усугубить нефропатию мочевой кислоты, в противном случае, вызванная гиперлактатемией. В подростковом возрасте и за его пределами заболевание клубочков может независимо развиваться, изначально представляя в виде клубочковой гиперфильтрации, указанной повышенным мочевым EGFR.
Спленомегалия
[ редактировать ]Увеличение селезенки (спленомегалия) распространено в GSD I и имеет две основные причины. В GSD IA спленомегалия может быть вызвана отношением между печени и селезенкой, которая заставляет либо расти, либо уменьшаться, чтобы соответствовать относительному размеру другого, в уменьшении степени. В GSD IB это побочный эффект использования Filgrastim для лечения нейтропении.
Эффекты кишечника
[ редактировать ]Вовлечение кишечника может вызвать легкую мультиногубку с жирным стулом ( стеаторея ), но обычно не требует лечения.
Риск заражения
[ редактировать ]Нейтропения является отличительной чертой GSD IB, отсутствующей в GSD IA. Микробиологическая причина нейтропении в GSD IB не совсем понятна. В целом, проблема возникает из -за нарушения клеточного метаболизма в нейтрофиле, что приводит к ускоренному апоптозу нейтрофилов. Нейтропения в GSD характеризуется как уменьшением абсолютного количества нейтрофилов, так и снижением функции нейтрофилов. Нейтрофилы используют специфический метаболический путь G6P, который опирается на присутствие G6Pase-β или G6PT для поддержания энергетического гомеостаза в клетке. Отсутствие G6PT в GSD IB ограничивает этот путь, что приводит к стрессу эндоплазматического ретикулума , окислительному стрессу в нейтрофиле, вызывающий преждевременный апоптоз. [ 3 ] Гранулоцитарные колони-стимулирующие фактор (G-CSF), доступный в качестве Filgrastim , может снизить риск инфекции. В некоторых случаях G-CSF, сформулированный как pegfilgrastim , проданный под торговым названием Neulasta, может использоваться в качестве альтернативы медленного действия, требуя менее частых дозирования.
Тромбоцитопения и проблемы с свертыванием крови
[ редактировать ]Нарушение агрегации тромбоцитов является необычным следствием хронической гипогликемии, наблюдаемой у пациентов с GSD I. Исследования продемонстрировали снижение функции тромбоцитов, характеризующееся снижением потребления протромбина , аномальных реакций агрегации, длительного времени кровотечения и низкой адгезивности тромбоцитов. Тяжесть дисфункции тромбоцитов обычно коррелирует с клиническими заболеваниями, причем наиболее тяжелые случаи коррелируют с лакциноцидозом и серьезной липидемией. [ 25 ] Это может вызвать клинически значимое кровотечение, особенно эпистаксис . Кроме того, пациенты с GSD I могут иметь тромбоцитопению в результате спленомегалии. В обстановке спленомегалии различные гематологические факторы могут быть секвестрированы в тканях селезенки, когда кровь фильтруется через орган. Это может снизить уровень тромбоцитов , доступных в кровотоке, что приводит к тромбоцитопении .
Эффекты развития
[ редактировать ]Задержка развития является потенциальным вторичным эффектом хронической или рецидивирующей гипогликемии, но, по крайней мере, теоретически предотвратима. Нормальные нейрональные и мышечные клетки не экспрессируют глюкозу-6-фосфатазу и, таким образом, не влияют GSD I напрямую. Однако без надлежащего лечения гипогликемии недостаток роста обычно является результатом хронически низкого уровня инсулина, персистирующего ацидоза, хронического повышения катаболических гормонов и калорийной недостаточности (или мультибсорбции ). Наиболее драматические задержки в развитии часто являются причиной серьезных (не только постоянных) эпизодов гипогликемии.
Диагноз
[ редактировать ]Несколько различных проблем могут привести к диагностике, обычно к двум годам:
- судороги или другие проявления тяжелой гипогликемии голодания
- гепатомегалия с брюшным выпуклом
- гипервентиляция и кажущаяся респираторная дистресс из -за метаболического ацидоза
- Эпизоды рвоты из -за метаболического ацидоза, часто ускоряемых незначительными заболеваниями и сопровождаются гипогликемией
Как только диагноз подозревается, множественность клинических и лабораторных признаков, как правило, делает сильный косвенный случай. Если гепатомегалия, гипогликемия натощак и плохой рост сопровождаются молочным ацидозом, гиперурикемией, гипертриглицеридемией и увеличенными почками с помощью ультразвука, GSD I является наиболее вероятной диагностикой. Список дифференциальных диагнозов включает в себя гликогенозы типов III и VI, дефицит фруктозы 1,6-бисфосфатазы и несколько других состояний (стр. 5) [ Цитация необходима ] , но никто не может произвести все особенности GSD I.
Следующим шагом обычно тщательно контролируется быстро. Гипогликемия часто происходит в течение шести часов. Критический образец крови, полученный во время гипогликемии, обычно выявляет умеренный метаболический ацидоз, высокие свободные жирные кислоты и бетагидроксибутират, очень низкие уровни инсулина и высокий уровень глюкагона, кортизола и гормона роста. Введение внутримышечного или внутривенного глюкагона (от 0,25 до 1 мг, в зависимости от возраста) или адреналина производит небольшой рост сахара в крови.
Диагноз окончательно подтверждается биопсией печени с помощью электронной микроскопии и анализа активности глюкозо-6-фосфатазы в ткани и/или специфических генных тестирования, доступных в последние годы.
Уход
[ редактировать ]Основной целью лечения является профилактика гипогликемии и вторичные метаболические расстройства частыми кормлениями продуктов с высоким содержанием глюкозы или крахмала (который легко расщепляется в глюкозу). Чтобы компенсировать неспособность печени обеспечить сахар, общее количество диетического углевода должно приближаться к 24-часовой скорости производства глюкозы. Диета должна содержать приблизительно 65–70% углеводов, 10–15% белка и 20–25% жира. По крайней мере, треть углеводов должны поставляться в течение ночи, так что маленький ребенок идет не более 3–4 часов без потребления углеводов. После того, как диагноз поставлен, приоритетом в лечении GSD I является поддержание адекватной глюкозы в крови. Пациенты стремятся поддерживать глюкозу в крови выше отсечения 72 мг/дл (4,0 ммоль/л) для гипогликемии. У пациентов с GSD IB есть дополнительный приоритет лечения, относящиеся к нейтропении. Правильное лечение глюкозы в крови в GSD I имеет решающее значение для предотвращения более серьезных эффектов высокого уровня молочной кислоты и мочевая кислота в крови и развитие печеночных аденом .
За последние 30 лет для достижения этой цели у маленьких детей были использованы два метода: (1) непрерывная инфузия глюкозы или крахмала глюкозы или крахмала; и (2) ночные кормления сырого кукурузного крахмала. Элементная формула, глюкозовый полимер и/или кукурузный крахмал можно непрерывно внедрять в течение ночи со скоростью, поставляющей 0,5–0,6 г/ч/ч глюкозы для младенца, или 0,3–0,4 для старшего ребенка. Этот метод требует назогастральной или гастростомической трубки и насоса. Внезапная смерть от гипогликемии произошла из -за неисправности или разъединения, а периодические кормления кукурузного крахмала теперь предпочтительнее непрерывной инфузии.
Кукурузный крахмал - это недорогой способ обеспечения постепенно перевариваемой глюкозы. Одна столовая ложка содержит почти 9 г углеводов (36 калорий). Хотя это безопаснее, дешевле и не требует оборудования, этот метод требует, чтобы родители возникали каждые 3–4 часа для управления кукурузным крахмалом. Типичное требование для маленького ребенка составляет 1,6 г/кг каждые 4 часа.
Долгосрочное лечение должно устранить симптомы гипогликемии и поддерживать нормальный рост. Обработка должна достигать нормального уровня глюкозы, молочной кислоты и электролита, а также только легкое повышение мочевой кислоты и триглицеридов.
Избегание других сахаров
[ редактировать ]Потребление углеводов , которые должны быть преобразованы в G6P для использования (например, галактоза и фруктоза ), должно быть сведено к минимуму. Хотя элементарные формулы доступны для детей, многие продукты содержат фруктозу или галактозу в формах сахарозы или лактозы . Приверженность становится спорной проблемой лечения после младенчества.
Другие терапевтические меры
[ редактировать ]Постоянное повышение мочевой кислоты выше 6,5 мг/дл требует лечения аллопуринолом для предотвращения осаждения мочевой кислоты в почках и суставах.
Из -за потенциала для нарушения функции тромбоцитов следует проверить способность коагуляции, а метаболическое состояние нормализовано перед операцией. Время кровотечения может быть нормализовано с 1-2 днями нагрузки глюкозы и улучшено с помощью DDAVP. Во время операции внутривенные жидкости должны содержать 10% декстрозы и без лактата.
Пациент с GSD, тип 1B лечился пересадкой печени в медицинском центре UCSF в 1993 году, что привело к разрешению гипогликемических эпизодов и необходимости того, чтобы пациент держался подальше от природных источников сахара. Другие пациенты также прошли эту процедуру с положительными результатами. Хотя пересадка печени привела к разрешению гипогликемии, она, однако, не разрешила хроническую нейтропению и риск инфекции у пациентов.
Лечение эпизодов острого метаболического ацидоза
[ редактировать ]Наиболее значительной острой проблемой в детстве является уязвимость к эпизодам метаболического ацидоза, ускоренного незначительными заболеваниями. Если рвотная болезнь сохраняется дольше 2–4 часов, ребенка следует увидеть и оценивать для обезвоживания, ацидоза и гипогликемии. Если они развиваются, внутривенные жидкости должны быть предоставлены со скоростью технического обслуживания. Для мягкого ацидоза эффективная жидкость составляет 10% декстрозы в ½ нормального физиологического раствора с 20 мг -экв/л KCl, но если ацидоз тяжелый, 75–100 мг -экв/л Nahco
3 и 20 мэкв/л ацетата могут быть заменены NaCl и KCl.
Метаболический контроль
[ редактировать ]Метаболический контроль часто уменьшается во время и после полового созревания, в результате того, что пациент перерастает свой план диетического лечения. [ 26 ]
Прогноз
[ редактировать ]Без адекватного метаболического лечения пациенты с GSD я умер в младенчестве или в детстве от подавляющей гипогликемии и ацидоза. Те, кто выжил, были задержлены в физическом росте и задержаны в половой зрелости из -за хронически низкого уровня инсулина. Интеллектуальная инвалидность, возникающая в результате рецидивирующей, тяжелой гипогликемии, считается предотвратимой при надлежащем лечении.
Осложнения в печени были серьезными у некоторых пациентов. Аденомы печени могут развиваться во втором десятилетии или позже, с небольшими шансами на позднюю злокачественную трансформацию в гепатому или печеночную карциномы (обнаруживается при скрининге альфа-фетопротеина). Несколько детей с продвинутыми осложнениями в печени улучшились после трансплантации печени.
Дополнительные проблемы, зарегистрированные у подростков и взрослых с GSD, я включил гиперурикемическую подагра , панкреатит и хроническую почечную недостаточность . Несмотря на гиперлипидемию , атеросклеротические осложнения редки.
С диагнозом до возникновения серьезного вреда, быстрого изменения ацидотических эпизодов и соответствующего долгосрочного лечения, большинство детей будут здоровыми. За исключением и квалификацией, здоровье и продолжительность жизни взрослых также могут быть довольно хорошими, хотя отсутствие эффективного лечения до середины 1980-х годов означает, что информация о долгосрочной эффективности ограничена.
Эпидемиология
[ редактировать ]В Соединенных Штатах GSD I имеет частоту примерно 1 из 50 000 [ 27 ] до 100 000 [ 28 ] рождение. Ни один из гликогенозов в настоящее время не обнаруживается стандартным или расширенным скринингом новорожденных .
Болезнь чаще встречается у людей ашкенази -еврейского , мексиканского, китайского и японского происхождения. [ 29 ]
Ссылки
[ редактировать ]- ^ Вейга-да-кунья, М.; Герин, я.; Ван Шафтинген, Э. (май 2000). "Сколько форм гликогенового хранения болезней типа I?" Полем Европейский журнал педиатрии . 159 (5): 314–318. doi : 10.1007/s004310051279 . ISSN 0340-6199 . PMID 10834514 .
- ^ Jump up to: а беременный Носимен, Джейкоб М.; Мисдраджи, Джозеф; Кори, Кэтлин Э. (май 2012 г.). «Вторичные причины неалкогольной жировой болезни печени» . Терапевтические достижения в области гастроэнтерологии . 5 (3): 199–207. doi : 10.1177/1756283x11430859 . ISSN 1756-283X . PMC 3342568 . PMID 22570680 .
- ^ Jump up to: а беременный в дюймовый Чоу, Дженис Ю.; Джун, Хён Сик; Мэнсфилд, Брайан С. (январь 2010 г.). «Нейтропения при заболевании гликогена типа IB» . Современное мнение о гематологии . 17 (1): 36–42. doi : 10.1097/moh.0b013e328331df85 . ISSN 1065-6251 . PMC 3099242 . PMID 19741523 .
- ^ Jump up to: а беременный Дейл, Дэвид С.; Болярд, Одри Анна; Марреро, Трейси М.; Фан, Лан; Боксер, Лоуренс А.; Кишнани, Прия С.; Курцберг, Джоан; Вайнштейн, Дэвид А. (2011-11-18). «Нейтропения при хранении гликогена 1B (GSD1B)». Кровь . 118 (21): 4791. doi : 10.1182/blood.v118.21.4791.4791 . ISSN 0006-4971 .
- ^ Виссер, Гепке; Рейбл, Ян Питер; Лабрун, Филипп; Леонард, Джеймс В.; Моисей, Симон; Улрих, Курт; Вендель, Удо; Groenier, Klaas H.; Смит, Г. Питер А. (октябрь 2002 г.). «Гранулоцитарные колони-стимулирующие фактор в гликогенном хранении заболевания типа 1B. Европейский журнал педиатрии . 161 (Suppl 1): S83–87. doi : 10.1007/s00431-002-1010-0 . ISSN 0340-6199 . PMID 12373578 . S2CID 33840498 .
- ^ «Гликогеное хранение типа I типа I» . Норд (Национальная организация редких расстройств) . Получено 2019-09-29 .
- ^ Синдром Гирке в том, кто его назвал?
- ^ Гирке, Э. (1929). «Гепато-нефромегалия Glycogenica (болезнь хранения гликогена в печени и почках)». Вклад в патологическую анатомию и общую патологию . 82 Йена: 497–513.
- ^ Парих, Нирзар С.; Ahlawat, Rajni (2019), «Гликогеное хранение болезни I типа I (болезнь фон Гирке)» , Statpearls , Statpearls Publishing, PMID 30480935 , получен 2019-11-01
- ^ Джун, Хён Сик; Вайнштейн, Дэвид А.; Ли, молодой мок; Мэнсфилд, Брайан С.; Чоу, Дженис Ю. (2014-05-01). «Молекулярные механизмы дисфункции нейтрофилов при хранении гликогена типа IB типа IB» . Кровь . 123 (18): 2843–2853. doi : 10.1182/blood-2013-05-502435 . ISSN 0006-4971 . PMC 4007611 . PMID 24565827 .
- ^ Jump up to: а беременный Кишнани, Прия С.; Остин, Стефани Л.; Abdenur, Jose E.; Арн, Памела; Бали, Дикша С.; Бони, Энн; Чунг, Венди К.; Дагли, Адити I.; Дейл, Дэвид; Кеберл, Дуайт; Сомерс, Майкл Дж. (Ноябрь 2014 г.). «Диагностика и лечение гликогена хранения болезней I типа I: практическое руководство Американского колледжа медицинской генетики и геномики» . Генетика в медицине . 16 (11): E1. doi : 10.1038/gim.2014.128 . ISSN 1530-0366 . PMID 25356975 .
- ^ Labrune, P.; Trioche, P.; Duvaltier, я.; Chevalier, P.; Odièvre, M. (март 1997 г.). «Гепатоцеллюлярные аденомы в гликогенном хранении типа I и III: серия из 43 пациентов и обзор литературы» . Журнал педиатрической гастроэнтерологии и питания . 24 (3): 276–279. doi : 10.1097/00005176-199703000-00008 . ISSN 0277-2116 . PMID 9138172 .
- ^ Jump up to: а беременный Ван, Дэвид Q.; Carreras, Caroline T.; Фиске, Лори М.; Остин, Стефани; Бори, Даниэль; Кишнани, Прия С.; Вайнштейн, Дэвид А. (сентябрь 2012 г.). «Характеристика и патогенез анемии при хранении гликогена типа IA и IB» . Генетика в медицине . 14 (9): 795–799. doi : 10.1038/gim.2012.41 . ISSN 1098-3600 . PMC 3808879 . PMID 22678084 .
- ^ Дэвид, Вайнштейн (2019-09-04). «Ultragenyx DTX401 Фаза 1/2 COHORT 2 Conference Conference» . Edge.Media-server.com . Получено 2019-10-30 .
- ^ Такамацу, Ясуши; Джими, Широ; Сато, Томохито; Хара, Шууджи; Suzumiya, Junji; Тамура, Казуо (январь 2007 г.). «Тромбоцитопения в связи со спленомегалией во время лечения гранулоцитов-колони-стимулирующим фактором у мышей не вызвана гиперспленизмом и разрешено спонтанно» . Переливание . 47 (1): 41–49. doi : 10.1111/j.1537-2995.2007.01061.x . ISSN 0041-1132 . PMID 17207228 . S2CID 36128181 .
- ^ Чен, Цзы-Лин; Чиан, Я-Вэнь; Лин, Гуаньли; Чанг, Син-Хоу; Удержание, Те-Шенг; Шех, Мин Хуа; Sun, Der-Shan (2018-05-02). «Различные эффекты гранулоцитарного колонимулирующего фактора и эритропоэтина на эритропоэз» . Исследование стволовых клеток и терапия . 9 (1): 119. doi : 10.1186/s13287-018-0877-2 . ISSN 1757-6512 . PMC 5930863 . PMID 29720275 .
- ^ «Гликогеное хранение типа I типа I» . Генетическая домашняя ссылка Национальной библиотеки медицины США и Национальных институтов здравоохранения . Получено 6 июля 2013 года .
- ^ Jump up to: а беременный Бали, DS; Чен, YT; Гольдштейн, JL; Булавка, Ра; Адам, депутат; Птица, ТД; Долан, кр; Fong, Ct; и др. (1993). «Гликогеное хранение типа I типа I». PMID 20301489 .
{{cite journal}}: CITE Journal требует|journal=( помощь ) - ^ Онлайн -наследование Менделя в человеке (OMIM): Заболевание гликогена IB - 232220
- ^ Онлайн -наследование Менделя в человеке (OMIM): Гликогеное хранение заболевания IC - 232240
- ^ Паркер, Пол (1981). «Регрессия печеночных аденом при хранении гликогена типа Ia с диетической терапией» . Гастроэнтерология . 81 (3): 534–536. doi : 10.1016/0016-5085 (81) 90606-5 . PMID 6941908 .
- ^ Минарих, Лори А.; Кирпич, Александр; Фиске, Лори М.; Вайнштейн, Дэвид А. (2013). «Минеральная плотность кости в гликогенном хранении типа IA и IB» . Генетика в медицине . 14 (8): 737–741. doi : 10.1038/gim.2012.36 . ISSN 1098-3600 . PMC 3884026 . PMID 22481133 .
- ^ Soejima, K.; Посадка, BH; ROE, TF; Swanson, VL (1985). «Патологические исследования остеопороза болезни фон Гирке (гликогеноз 1а)». Педиатрическая патология . 3 (2–4): 307–319. doi : 10.3109/15513818509078791 . ISSN 0277-0938 . PMID 3867867 .
- ^ Пан, Бо-Лин; Локе, Song-Seng (2018-01-10). «Хроническое заболевание почек, связанное со снижением минеральной плотности кости, мочевой кислотой и метаболическим синдромом» . Plos один . 13 (1): E0190985. BIBCODE : 2018PLOSO..1390985P . doi : 10.1371/journal.pone.0190985 . ISSN 1932-6203 . PMC 5761949 . PMID 29320555 .
- ^ Czapek, Emily E.; Дейкин, Даниэль; Salzman, Edwin W. (1973-02-01). «Дисфункция тромбоцитов при хранении гликогена типа I типа I» . Кровь . 41 (2): 235–247. doi : 10.1182/blood.v41.2.235.235 . ISSN 0006-4971 . PMID 4350560 .
- ^ Деркс, Терри Г.Дж; Van Rijn, Margreet (2015). «Липиды при заболеваниях хранения гликогена печени: патофизиология, мониторинг управления питанием и будущих направлений» . Журнал наследственного метаболического заболевания . 38 (3): 537–543. doi : 10.1007/s10545-015-9811-2 . ISSN 0141-8955 . PMC 4432100 . PMID 25633903 .
- ^ Болезнь хранения гликогена типа I в эмедицине
- ^ https://rarediseases.org/rare-diseases/glycogen-storage-disease-type-i/ Национальная организация для редких расстройств
- ^ Голдман, Ли (2011). Сесил Медицина Голдмана (24 -е изд.). Филадельфия: Elsevier Saunders. с. 1356 . ISBN 978-1437727883 .
Дальнейшее чтение
[ редактировать ]Внешние ссылки
[ редактировать ] СМИ, связанные с гликогеном хранения, тип типа I в Wikimedia Commons
СМИ, связанные с гликогеном хранения, тип типа I в Wikimedia Commons