Белковый дизайн
Дизайн белка — это рациональный дизайн новых белковых молекул для создания новой активности, поведения или цели, а также для продвижения базового понимания функции белка. [1] Белки можно создавать с нуля ( дизайн de novo ) или путем создания расчетных вариантов известной структуры белка и ее последовательности (так называемый редизайн белка ). Подходы к рациональному проектированию белков позволяют прогнозировать белковые последовательности, которые будут складываться в определенные структуры. Эти предсказанные последовательности затем могут быть проверены экспериментально с помощью таких методов, как синтез пептидов , сайт-направленный мутагенез или синтез искусственных генов .
Рациональный дизайн белка появился в середине 1970-х годов. [2] Однако в последнее время появилось множество примеров успешного рационального проектирования водорастворимых и даже трансмембранных пептидов и белков, отчасти благодаря лучшему пониманию различных факторов, способствующих стабильности структуры белка , и разработке более совершенных вычислительных методов.
Обзор и история
[ редактировать ]Целью рационального проектирования белков является предсказание аминокислотных последовательностей , которые будут складываться в определенную структуру белка. Хотя количество возможных белковых последовательностей огромно и растет экспоненциально с размером белковой цепи, только часть из них надежно и быстро сворачивается в одно нативное состояние . Дизайн белка включает идентификацию новых последовательностей в этом подмножестве. Нативное состояние белка — это минимум конформационной свободной энергии цепи. Таким образом, дизайн белка – это поиск последовательностей, имеющих выбранную структуру как минимум свободной энергии. В каком-то смысле это противоположно предсказанию структуры белка . При проектировании указывается третичная структура и определяется последовательность, которая будет сворачиваться в нее. Следовательно, это также называется обратным складыванием . В таком случае дизайн белка представляет собой проблему оптимизации: с использованием некоторых критериев оценки выбирается оптимизированная последовательность, которая будет складываться до желаемой структуры.
Когда в 1970-х и 1980-х годах были рационально разработаны первые белки, их последовательность оптимизировалась вручную на основе анализа других известных белков, состава последовательностей, зарядов аминокислот и геометрии желаемой структуры. [2] Первые разработанные белки приписываются Бернду Гутте, который разработал уменьшенную версию известного катализатора, бычьей рибонуклеазы, и третичные структуры, состоящие из бета-листов и альфа-спиралей, включая связующее вещество ДДТ . Позже Урри и его коллеги разработали эластин -подобные волокнистые пептиды, основываясь на правилах композиции последовательностей. Ричардсон и его коллеги разработали белок из 79 остатков, не имеющий гомологии последовательности с известным белком. [2] В 1990-х годах появление мощных компьютеров, библиотек конформаций аминокислот и силовых полей, разработанных в основном для молекулярно-динамического моделирования, позволило разработать инструменты вычислительного проектирования белков на основе структуры. После разработки этих вычислительных инструментов за последние 30 лет был достигнут большой успех в проектировании белков. Первый белок, успешно разработанный полностью de novo, был создан Стивеном Мэйо и его коллегами в 1997 году. [3] и вскоре после этого, в 1999 году, Питер С. Ким и его коллеги разработали димеры, тримеры и тетрамеры неестественных правозакрученных спиралей . [4] [5] В 2003 году лаборатория Дэвида Бейкера разработала полноценный белок со структурой, никогда ранее не встречавшейся в природе. [6] Позже, в 2008 году, группа Бейкера с помощью вычислений разработала ферменты для двух разных реакций. [7] В 2010 году одно из самых мощных нейтрализующих антител широкого спектра действия было выделено из сыворотки пациентов с помощью белкового зонда, разработанного с помощью вычислений. [8] Благодаря этим и другим успехам (см. примеры ниже), дизайн белка стал одним из наиболее важных инструментов, доступных для белковой инженерии . Есть большая надежда, что создание новых белков, малых и больших, найдет применение в биомедицине и биоинженерии .
Основные модели структуры и функции белка
[ редактировать ]Программы проектирования белков используют компьютерные модели молекулярных сил, которые управляют белками в среде in vivo . Чтобы сделать проблему разрешимой, эти силы упрощаются с помощью моделей белкового дизайна. Хотя программы проектирования белков сильно различаются, они должны решать четыре основных вопроса моделирования: какова целевая структура дизайна, какая гибкость допускается в целевой структуре, какие последовательности включены в поиск и какое силовое поле будет использоваться для последовательности и структуры оценок.
Целевая структура
[ редактировать ]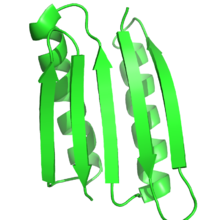
Функция белка во многом зависит от структуры белка, и рациональный дизайн белков использует эту взаимосвязь для проектирования функций путем создания белков, которые имеют целевую структуру или складку. Таким образом, по определению, при рациональном проектировании белка целевая структура или ансамбль структур должны быть известны заранее. Это контрастирует с другими формами белковой инженерии, такими как направленная эволюция , где используются различные методы для поиска белков, выполняющих определенную функцию, и с предсказанием структуры белка , когда последовательность известна, но структура неизвестна.
Чаще всего целевая структура основана на известной структуре другого белка. Однако новые складки, не встречающиеся в природе, становятся все более возможными. Питер С. Ким и его коллеги разработали тримеры и тетрамеры неестественных закрученных спиралей, которые раньше не встречались в природе. [4] [5] Белок Top7, разработанный в лаборатории Дэвида Бейкера , был полностью разработан с использованием алгоритмов проектирования белков и имел совершенно новую структуру. [6] Совсем недавно Бейкер и его коллеги разработали ряд принципов создания идеальных структур глобулярных белков на основе воронок сворачивания белков , которые служат мостом между предсказанием вторичной структуры и третичными структурами. Эти принципы, основанные как на предсказании структуры белка, так и на его проектировании, были использованы для разработки пяти различных новых топологий белка. [9]
Пространство последовательности
[ редактировать ]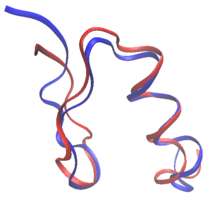
При рациональном проектировании белков белки могут быть переработаны на основе последовательности и структуры известного белка или полностью с нуля при de novo проектировании белков . При редизайне белка большинство остатков в последовательности сохраняются как аминокислоты дикого типа, в то время как некоторым разрешается мутировать. При проектировании de novo вся последовательность создается заново, без какой-либо предшествующей последовательности.
Как дизайн de novo , так и редизайн белка могут установить правила в пространстве последовательностей : конкретные аминокислоты, которые разрешены в каждом положении мутабельного остатка. Например, состав поверхности зонда RSC3 для отбора антител, широко нейтрализующих ВИЧ, был ограничен на основании эволюционных данных и баланса зарядов. Многие из самых ранних попыток создания белков в значительной степени основывались на эмпирических правилах пространства последовательностей. [2] Более того, дизайн фиброзных белков обычно следует строгим правилам в отношении пространства последовательностей. коллагена , часто состоят из повторяющихся структур Gly-Pro-X. Например, белки, созданные на основе [2] Появление вычислительных методов позволяет создавать белки без вмешательства человека в выбор последовательностей. [3]
Структурная гибкость
[ редактировать ]
При проектировании белка известна целевая структура (или структуры) белка. Однако подход к рациональному проектированию белков должен моделировать некоторую гибкость целевой структуры, чтобы увеличить количество последовательностей, которые можно спроектировать для этой структуры, и минимизировать вероятность сворачивания последовательности в другую структуру. Например, при редизайне белка одной небольшой аминокислоты (такой как аланин) в плотно упакованном ядре белка с помощью рационального подхода к проектированию можно было бы предсказать, что очень немногие мутанты свернутся до целевой структуры, если окружающие боковые цепи переупаковке не подлежат.
Таким образом, важным параметром любого процесса проектирования является степень гибкости, допускаемая как для боковых цепей, так и для магистральной цепи. В простейших моделях основная цепь белка остается жесткой, в то время как некоторым боковым цепям белка разрешено изменять конформацию. Однако боковые цепи могут иметь много степеней свободы в отношении длины связей, валентных углов и χ двугранных углов . Чтобы упростить это пространство, в методах проектирования белков используются библиотеки ротамеров, которые предполагают идеальные значения длин связей и валентных углов, ограничивая при этом двугранные углы χ несколькими часто наблюдаемыми низкоэнергетическими конформациями, называемыми ротамерами .
Библиотеки ротамеров получены в результате статистического анализа многих белковых структур. Независимые от основной цепи библиотеки ротамеров описывают все ротамеры. [10] Библиотеки ротамеров, зависимые от основной цепи , напротив, описывают ротамеры как вероятность их появления в зависимости от расположения основной цепи белка вокруг боковой цепи. [11] Большинство программ проектирования белков используют одну конформацию (например, модальное значение для двугранников ротамера в пространстве) или несколько точек в области, описываемой ротамером; программа проектирования белков OSPREY, напротив, моделирует всю непрерывную область. [12]
Хотя рациональный дизайн белка должен сохранять общую складку основной цепи белка, обеспечение некоторой гибкости основной цепи может значительно увеличить количество последовательностей, которые сворачиваются в структуру, сохраняя при этом общую складку белка. [13] Гибкость остова особенно важна при редизайне белка, поскольку мутации последовательности часто приводят к небольшим изменениям в структуре остова. Более того, гибкость основной цепи может иметь важное значение для более продвинутых приложений проектирования белков, таких как прогнозирование связывания и разработка ферментов. Некоторые модели гибкости основной цепи белкового дизайна включают небольшие и непрерывные глобальные движения основной цепи, дискретные образцы основной цепи вокруг целевой складки, растирающие движения и гибкость белковой петли. [13] [14]
Энергетическая функция
[ редактировать ]
Методы рационального проектирования белков должны быть способны отличать последовательности, которые будут стабильны под целевой укладкой, от тех, которые предпочли бы другие конкурирующие состояния с низкой энергией. Таким образом, дизайн белка требует точных энергетических функций , которые могут ранжировать и оценивать последовательности по тому, насколько хорошо они складываются в целевую структуру. В то же время, однако, эти энергетические функции должны учитывать вычислительные проблемы, стоящие за дизайном белка. Одним из самых сложных требований для успешного проектирования является то, чтобы энергетическая функция была одновременно точной и простой для вычислительных расчетов.
Наиболее точные энергетические функции основаны на квантово-механическом моделировании. Однако такое моделирование слишком медленное и обычно непрактично для проектирования белков. Вместо этого многие алгоритмы проектирования белков используют либо энергетические функции, основанные на физике, адаптированные из молекулярной механики программ моделирования , либо энергетические функции, основанные на знаниях , или гибридное сочетание того и другого. Тенденция заключалась в использовании большего количества функций потенциальной энергии, основанных на физике. [15]
Энергетические функции, основанные на физике, такие как AMBER и CHARMM , обычно получаются на основе квантово-механического моделирования и экспериментальных данных термодинамики, кристаллографии и спектроскопии. [16] Эти энергетические функции обычно упрощают функции физической энергии и делают их попарно разлагаемыми, а это означает, что полную энергию конформации белка можно рассчитать путем сложения попарной энергии между каждой парой атомов, что делает их привлекательными для алгоритмов оптимизации. Энергетические функции, основанные на физике, обычно моделируют термин Леннарда-Джонса притяжения-отталкивания между атомами и парный электростатики. кулоновский член [17] между несвязанными атомами.

Статистические потенциалы, в отличие от потенциалов, основанных на физике, имеют то преимущество, что их можно быстро вычислить, неявно учитывать сложные эффекты и они менее чувствительны к небольшим изменениям в структуре белка. [19] Эти энергетические функции основаны на получении значений энергии на основе частоты появления в структурной базе данных.
Однако к дизайну белков предъявляются требования, которые иногда могут быть ограничены силовыми полями молекулярной механики. Силовые поля молекулярной механики, которые были используемые в основном при моделировании молекулярной динамики, оптимизированы для моделирования отдельных последовательностей, но при проектировании белков осуществляется поиск множества конформаций многих последовательностей. Таким образом, силовые поля молекулярной механики должны быть адаптированы для проектирования белков. На практике энергетические функции проектирования белков часто включают в себя как статистические термины, так и термины, основанные на физике. Например, энергетическая функция Розетты, одна из наиболее часто используемых энергетических функций, включает в себя энергетические термины, основанные на физике, происходящие из энергетической функции CHARMM, и статистические энергетические термины, такие как вероятность ротамера и электростатика, основанная на знаниях. Как правило, энергетические функции индивидуально настраиваются между лабораториями и специально адаптируются для каждого проекта. [16]
Проблемы эффективного проектирования энергетических функций
[ редактировать ]Вода составляет большую часть молекул, окружающих белки, и является основной движущей силой структуры белка. Таким образом, моделирование взаимодействия воды и белка имеет жизненно важное значение для проектирования белков. Число молекул воды, которые взаимодействуют с белком в любой момент времени, огромно, и каждая из них имеет большое количество степеней свободы и партнеров по взаимодействию. Вместо этого программы проектирования белков моделируют большинство таких молекул воды как континуум, моделируя как гидрофобный эффект, так и сольватационную поляризацию. [16]
Отдельные молекулы воды иногда могут играть решающую структурную роль в ядре белков, а также во взаимодействиях белок-белок или белок-лиганд. Неспособность смоделировать такие воды может привести к неправильному прогнозированию оптимальной последовательности межбелкового интерфейса. В качестве альтернативы к ротамерам можно добавить молекулы воды. [16]
Как задача оптимизации
[ редактировать ]
Цель дизайна белка — найти белковую последовательность, которая будет сворачиваться до целевой структуры. Таким образом, алгоритм проектирования белка должен искать все конформации каждой последовательности относительно целевой складки и ранжировать последовательности в соответствии с конформацией каждой из них с наименьшей энергией, как это определяется энергетической функцией проектирования белка. Таким образом, типичными входными данными для алгоритма проектирования белка являются целевая складка, пространство последовательностей, структурная гибкость и энергетическая функция, а выходными данными являются одна или несколько последовательностей, которые, по прогнозам, будут стабильно сворачиваться в целевую структуру.
Однако количество белковых последовательностей-кандидатов растет экспоненциально с увеличением количества белковых остатков; например, есть 20 100 белковые последовательности длиной 100. Более того, даже если конформации боковой цепи аминокислот ограничены несколькими ротамерами (см. Структурная гибкость ), это приводит к экспоненциальному числу конформаций для каждой последовательности. Таким образом, в нашем белке, состоящем из 100 остатков, и предполагая, что каждая аминокислота имеет ровно 10 ротамеров, алгоритм поиска, который ищет это пространство, должен будет искать более 200 остатков. 100 конформации белка.
Наиболее распространенные энергетические функции можно разложить на попарные члены между ротамерами и типами аминокислот, что делает проблему комбинаторной, и для ее решения можно использовать мощные алгоритмы оптимизации. В этих случаях полная энергия каждой конформации, принадлежащей каждой последовательности, может быть сформулирована как сумма индивидуальных и попарных членов между положениями остатков. Если дизайнера интересует только лучшая последовательность, алгоритм проектирования белка требует только конформации с наименьшей энергией последовательности с наименьшей энергией. В этих случаях аминокислотную идентичность каждого ротамера можно игнорировать и все ротамеры, принадлежащие к разным аминокислотам, можно рассматривать одинаково. Пусть r i — ротамер в положении остатка i в белковой цепи, а E( r i ) — потенциальная энергия между внутренними атомами ротамера. Пусть E ( r i , r j ) будет потенциальной энергией между r i и ротамером r j в положении остатка j . Затем мы определяем задачу оптимизации как задачу поиска конформации минимальной энергии ( И ) :
| ( 1 ) |
![{\displaystyle \min E_{T} =\sum _{i}{\Big [}E_{i}(r_{i})+\sum _{i\neq j}E_{ij}(r_{i} ,r_{j}){\Big ]}\,}](https://wikimedia.org/api/rest_v1/media/math/render/svg/3332d826843218136390cef20e4ee8e3694fc477)
Задача минимизации E T является NP-трудной задачей. [14] [20] [21] Несмотря на то, что класс задач является NP-сложным, на практике многие случаи проектирования белков могут быть точно решены или удовлетворительно оптимизированы с помощью эвристических методов.
Алгоритмы
[ редактировать ]Несколько алгоритмов были разработаны специально для задачи проектирования белков. Эти алгоритмы можно разделить на два широких класса: точные алгоритмы, такие как устранение тупиков , которые не имеют гарантий времени выполнения , но гарантируют качество решения; и эвристические алгоритмы, такие как Монте-Карло, которые работают быстрее, чем точные алгоритмы, но не имеют гарантий оптимальности результатов. Точные алгоритмы гарантируют, что процесс оптимизации дает оптимальные результаты в соответствии с моделью проектирования белка. Таким образом, если предсказания точных алгоритмов терпят неудачу, когда они подтверждаются экспериментально, то источником ошибки можно отнести энергетическую функцию, допустимую гибкость, пространство последовательностей или целевую структуру (например, если ее невозможно спроектировать). [22]
Некоторые алгоритмы проектирования белков перечислены ниже. Хотя эти алгоритмы решают только самую базовую формулировку проблемы проектирования белка, уравнение ( 1 ), когда цель оптимизации меняется, поскольку дизайнеры вносят улучшения и расширения в модель проектирования белка, такие как улучшения допустимой структурной гибкости (например, основная цепь белка гибкость) или включая сложные энергетические термины, многие расширения дизайна белков, улучшающие моделирование, построены на основе этих алгоритмов. Например, Rosetta Design включает сложные термины энергопотребления и гибкость магистральной сети, используя метод Монте-Карло в качестве основного алгоритма оптимизации. Алгоритмы OSPREY основаны на алгоритме устранения тупиков и A* для включения непрерывных движений основной и боковой цепи. Таким образом, эти алгоритмы дают хорошее представление о различных видах алгоритмов, доступных для проектирования белков.
В 2020 году ученые сообщили о разработке процесса на основе искусственного интеллекта с использованием баз данных генома для эволюционного проектирования новых белков. Они использовали глубокое обучение для определения правил проектирования. [23] [24] В 2022 году в исследовании сообщалось о программном обеспечении глубокого обучения, которое может создавать белки, содержащие заранее определенные функциональные сайты. [25] [26]
С математическими гарантиями
[ редактировать ]Устранение тупика
[ редактировать ]Алгоритм устранения тупика (DEE) итеративно уменьшает пространство поиска проблемы, удаляя ротамеры, которые, как можно доказуемо, не являются частью глобальной конформации с наименьшей энергией (GMEC). На каждой итерации алгоритм устранения тупика сравнивает все возможные пары ротамеров в каждой позиции остатка и удаляет каждый ротамер r' i, который, как можно показать, всегда имеет более высокую энергию, чем другой ротамер r i, и, таким образом, не является частью ГМЭК:

Другие мощные расширения алгоритма исключения тупиков включают критерий исключения пар и обобщенный критерий исключения тупика . Этот алгоритм также был расширен для работы с ротамерами непрерывного действия с доказуемыми гарантиями.
Хотя алгоритм устранения тупиков работает за полиномиальное время на каждой итерации, он не может гарантировать сходимость. Если после определенного количества итераций алгоритм исключения тупиков не удаляет больше ротамеров, то необходимо либо объединить ротамеры, либо использовать другой алгоритм поиска для поиска в оставшемся пространстве поиска. В таких случаях устранение тупика действует как алгоритм предварительной фильтрации для уменьшения пространства поиска, в то время как другие алгоритмы, такие как A*, Монте-Карло, линейное программирование или FASTER, используются для поиска в оставшемся пространстве поиска. [14]
Ветвь и граница
[ редактировать ]Конформационное пространство дизайна белка можно представить в виде дерева , где остатки белка упорядочены произвольным образом, причем дерево разветвляется у каждого из ротамеров в остатке. Алгоритмы ветвей и границ используют это представление для эффективного исследования дерева конформации: при каждом разветвлении алгоритмы ветвей и границ ограничивают конформационное пространство и исследуют только перспективные ветви. [14] [27] [28]
Популярным алгоритмом поиска при проектировании белков является алгоритм поиска A* . [14] [28] A* вычисляет нижнюю границу оценки для каждого частичного древовидного пути, который ограничивает нижнюю границу (с гарантиями) энергии каждого из расширенных ротамеров. Каждая частичная конформация добавляется в приоритетную очередь, и на каждой итерации частичный путь с нижней нижней границей извлекается из очереди и расширяется. Алгоритм останавливается после перечисления полной конформации и гарантирует, что конформация является оптимальной.
Оценка A* f в дизайне белка состоит из двух частей: f=g+h . g — точная энергия ротамеров, которые уже были отнесены к частичной конформации. h — нижняя граница энергии ротамеров, которые еще не определены. Каждый из них устроен следующим образом, где d — индекс последнего присвоенного остатка в частичной конформации.

![{\displaystyle h=\sum _{j=d+1}^{n}[\min _{r_{j}}(E(r_{j})+\sum _{i=1}^{d} E(r_{i},r_{j})+\sum _{k=j+1}^{n}\min _{r_{k}}E(r_{j},r_{k}))] }](https://wikimedia.org/api/rest_v1/media/math/render/svg/e143d714d94f81766d65c1ab49da42eeeed08b4a)
Целочисленное линейное программирование
[ редактировать ]Задачу оптимизации ( ET уравнение ( 1 )) можно легко сформулировать как целочисленную линейную программу (ILP). [29] Одна из наиболее мощных формулировок использует двоичные переменные для представления присутствия ротамера и ребер в конечном решении и ограничивает решение наличием ровно одного ротамера для каждого остатка и одного парного взаимодействия для каждой пары остатков:

ул.



Решатели ILP, такие как CPLEX , могут вычислить точное оптимальное решение для больших случаев проблем проектирования белков. Эти решатели используют с помощью линейного программирования релаксацию задачи , где q i и q ij могут принимать непрерывные значения, в сочетании с алгоритмом ветвей и разрезов для поиска оптимального решения только в небольшой части конформационного пространства. Было показано, что решатели ILP решают многие случаи проблемы размещения боковой цепи. [29]
Приближения к двойственному линейному программированию, основанные на передаче сообщений
[ редактировать ]Решатели ILP зависят от алгоритмов линейного программирования (LP), таких как симплексные или барьерные методы, для выполнения релаксации LP на каждой ветви. Эти алгоритмы LP были разработаны как методы оптимизации общего назначения и не оптимизированы для задачи проектирования белков (уравнение ( 1 )). Как следствие, релаксация ЛП становится узким местом решателей ILP, когда размер задачи велик. [30] Недавно было разработано несколько альтернатив, основанных на алгоритмах передачи сообщений, специально для оптимизации LP-релаксации проблемы дизайна белка. Эти алгоритмы могут аппроксимировать как двойственный , так и простой пример целочисленного программирования, но для того, чтобы сохранить гарантии оптимальности, они наиболее полезны при использовании для аппроксимации двойственной задачи проектирования белка, поскольку аппроксимация двойственной задачи гарантирует отсутствие решений. пропущенный. Приближения, основанные на передаче сообщений, включают алгоритм передачи сообщений с максимальным произведением с перевзвешенным деревом , [31] [32] и алгоритм линейного программирования передачи сообщений . [33]
Алгоритмы оптимизации без гарантий
[ редактировать ]Монте-Карло и имитация отжига.
[ редактировать ]Монте-Карло — один из наиболее широко используемых алгоритмов проектирования белков. В своей простейшей форме алгоритм Монте-Карло выбирает остаток случайным образом, и в этом остатке оценивается случайно выбранный ротамер (любой аминокислоты). [21] Новая энергия белка E new сравнивается со старой энергией E old и новый ротамер принимается с вероятностью:

где β — постоянная Больцмана , а температуру T можно выбрать такой, чтобы на начальных этапах она была высокой и медленно отжигалась для преодоления локальных минимумов. [12]
БЫСТРЕЕ
[ редактировать ]Алгоритм FASTER использует комбинацию детерминированных и стохастических критериев для оптимизации аминокислотных последовательностей. FASTER сначала использует DEE для устранения ротамеров, которые не являются частью оптимального решения. Затем серия итеративных шагов оптимизирует назначение ротамера. [34] [35]
Распространение убеждений
[ редактировать ]При распространении убеждений при проектировании белка алгоритм обменивается сообщениями, описывающими убежденность каждого остатка в вероятности присутствия каждого ротамера в соседних остатках. Алгоритм обновляет сообщения на каждой итерации и выполняет итерации до сходимости или до фиксированного количества итераций. Конвергенция не гарантируется при проектировании белка. Сообщение m i→ j (r j , которое остаток i отправляет каждому ротамеру (r j в соседнем остатке j), определяется как:

Распространение убеждений как по максимальному, так и по суммарному произведению использовалось для оптимизации дизайна белка.
Приложения и примеры созданных белков
[ редактировать ]Ферментный дизайн
[ редактировать ]Создание новых ферментов — это использование дизайна белков с огромными биоинженерными и биомедицинскими применениями. В общем, проектирование структуры белка может отличаться от проектирования фермента, поскольку при проектировании ферментов необходимо учитывать множество состояний, участвующих в каталитическом механизме . Однако дизайн белка является предпосылкой дизайна ферментов de novo , потому что, по крайней мере, дизайн катализаторов требует каркаса, в который может быть встроен каталитический механизм. [36]
Большой прогресс в разработке и редизайне ферментов de novo был достигнут в первом десятилетии 21 века. В трех крупных исследованиях Дэвид Бейкер и его коллеги de novo разработали ферменты для ретроальдольной реакции . [37] реакция Кемпа-элиминирования, [38] и для реакции Дильса-Альдера . [39] Кроме того, Стивен Мэйо и его коллеги разработали итеративный метод разработки наиболее эффективного известного фермента для реакции элиминирования Кемпа. [40] Кроме того, в лаборатории Брюса Дональда компьютерный дизайн белка использовался для переключения специфичности одного из белковых доменов нерибосомальной пептидной синтетазы , которая продуцирует грамицидин S , с его природного субстрата фенилаланина на другие неродственные субстраты, включая заряженные аминокислоты; модифицированные ферменты имели активность, близкую к активности дикого типа. [41]
Полурациональный дизайн
[ редактировать ]Полурациональный дизайн — метод целенаправленной модификации, основанный на определенном понимании последовательности, структуры и каталитического механизма ферментов. Этот метод находится между иррациональным дизайном и рациональным дизайном. Он использует известную информацию и средства для выполнения эволюционной модификации конкретных функций целевого фермента. Характерной чертой полурационального замысла является то, что он не опирается исключительно на случайные мутации и скрининг, а сочетает в себе концепцию направленной эволюции. Он создает библиотеку случайных мутантов с разнообразными последовательностями посредством мутагенеза , подверженного ошибкам RCR , рекомбинации ДНК и мутагенеза с насыщением сайтов . В то же время он использует понимание ферментов и принципов проектирования для целенаправленного отбора мутантов с желаемыми характеристиками.
Методология полурационального дизайна подчеркивает глубокое понимание ферментов и контроль эволюционного процесса. Это позволяет исследователям использовать известную информацию для управления эволюционным процессом, тем самым повышая эффективность и уровень успеха. Этот метод играет важную роль в модификации функции белка, поскольку он может сочетать в себе преимущества иррационального и рационального дизайна, а также исследовать неизвестное пространство и использовать известные знания для целенаправленной модификации.
Полурациональный дизайн имеет широкий спектр применений, включая, помимо прочего, оптимизацию ферментов, модификацию мишеней лекарств, эволюцию биокатализаторов и т. д. С помощью этого метода исследователи могут более эффективно улучшать функциональные свойства белков для удовлетворения конкретных биотехнологических или медицинских требований. потребности. Хотя этот метод предъявляет высокие требования к информации и технологиям и сравнительно сложен в реализации, с развитием вычислительной техники и биоинформатики перспективы применения полурационального проектирования в белковой инженерии становятся все шире. [42]
Дизайн для близости
[ редактировать ]Белко-белковые взаимодействия участвуют в большинстве биотических процессов. Многие из наиболее трудно поддающихся лечению заболеваний, таких как болезнь Альцгеймера , многие формы рака (например, TP53 ) и инфекция вируса иммунодефицита человека ( ВИЧ ), связаны с белок-белковыми взаимодействиями. Таким образом, для лечения таких заболеваний желательно разработать белковые или белковоподобные терапевтические средства, которые связывают одного из партнеров взаимодействия и, таким образом, нарушают вызывающее заболевание взаимодействие. Это требует разработки белковой терапии, обеспечивающей сродство к своему партнеру.
Белково-белковые взаимодействия можно проектировать с использованием алгоритмов проектирования белков, поскольку принципы, определяющие стабильность белка, также управляют межбелковым связыванием. Однако проектирование белок-белкового взаимодействия сопряжено с проблемами, которые обычно не встречаются при проектировании белков. Одна из наиболее важных проблем заключается в том, что, как правило, границы раздела между белками более полярны, чем ядра белков, и связывание предполагает компромисс между десольватацией и образованием водородных связей. [43] Чтобы преодолеть эту проблему, Брюс Тидор и его коллеги разработали метод улучшения сродства антител, сосредоточив внимание на электростатическом вкладе. Они обнаружили, что для антител, разработанных в исследовании, снижение затрат на десольватацию остатков на границе раздела увеличивает аффинность связывающей пары. [43] [44] [45]
Оценка прогнозов привязки
[ редактировать ]поскольку связывание предполагает компромисс между конформациями с наименьшей энергией свободных белков ( EP для оценки прогнозов связывания , и EL Энергетические функции дизайна белка должны быть адаптированы ) и конформацией связанного комплекса с наименьшей энергией ( EP PL ):
- .

Алгоритм K* аппроксимирует константу связывания алгоритма, включая конформационную энтропию в расчет свободной энергии. Алгоритм K* рассматривает только конформации свободного и связанного комплексов с наименьшей энергией (обозначаемые множествами P , L и PL ) для аппроксимации статистических сумм каждого комплекса: [14]

Дизайн для специфики
[ редактировать ]Дизайн межбелковых взаимодействий должен быть высокоспецифичным, поскольку белки могут взаимодействовать с большим количеством белков; успешный дизайн требует отборных связующих. Таким образом, алгоритмы проектирования белков должны быть способны различать целевое (или позитивное проектирование ) и нецелевое связывание (или негативный дизайн ). [2] [43] Одним из наиболее ярких примеров разработки специфичности является разработка Эми Китинг и ее коллег специфических bZIP -связывающих пептидов для 19 из 20 семейств bZIP; 8 из этих пептидов были специфичнее к своему предполагаемому партнеру по сравнению с конкурирующими пептидами. [43] [46] [47] Кроме того, Андерсон и его коллеги также использовали позитивный и негативный дизайн для прогнозирования мутаций в активном сайте мишени лекарства, которые придают устойчивость к новому лекарству; положительный дизайн использовали для поддержания активности дикого типа, а отрицательный дизайн использовали для нарушения связывания лекарственного средства. [48] Недавняя модернизация вычислений, проведенная Костасом Маранасом и его коллегами, также позволила экспериментально переключить кофактора специфичность Candida boidinii ксилозоредуктазы с НАДФН на НАДН . [49]
Белковая шлифовка
[ редактировать ]Обновление поверхности белка заключается в моделировании поверхности белка при сохранении общей складки, ядра и пограничных областей белка нетронутыми. Обновление поверхности белка особенно полезно для изменения связывания белка с другими белками. Одним из наиболее важных применений обновления поверхности белка стала разработка зонда RSC3 для отбора широко нейтрализующих антител к ВИЧ в Исследовательском центре вакцин NIH. Во-первых, для конструирования были выбраны остатки за пределами границы связывания между белком оболочки ВИЧ gp120 и ранее обнаруженным b12-антителом. Затем последовательность была выбрана на основе эволюционной информации, растворимости, сходства с диким типом и других соображений. Затем программное обеспечение RosettaDesign использовалось для поиска оптимальных последовательностей в выбранном пространстве последовательностей. Позже RSC3 был использован для обнаружения широко нейтрализующего антитела VRC01 в сыворотке человека, длительно инфицированного ВИЧ и не прогрессирующего. [50]
Дизайн глобулярных белков
[ редактировать ]Глобулярные белки — это белки, которые содержат гидрофобное ядро и гидрофильную поверхность. Глобулярные белки часто принимают стабильную структуру, в отличие от волокнистых белков , которые имеют множественные конформации. Трехмерную структуру глобулярных белков обычно легче определить с помощью рентгеновской кристаллографии и ядерного магнитного резонанса, чем как волокнистых белков, так и мембранных белков , что делает глобулярные белки более привлекательными для проектирования белков, чем другие типы белков. Наиболее успешные разработки белков включали глобулярные белки. И RSD-1 , и Top7 представляли собой de novo конструкции глобулярных белков . Еще пять белковых структур были разработаны, синтезированы и проверены в 2012 году группой Бейкера. Эти новые белки не выполняют биотической функции, но их структуры призваны действовать как строительные блоки, которые можно расширить, включив в них функциональные активные центры. Структуры были найдены вычислительным путем с использованием новой эвристики, основанной на анализе соединительных петель между частями последовательности, которые определяют вторичные структуры. [51]
Дизайн мембранных белков
[ редактировать ]Было успешно создано несколько трансмембранных белков. [52] наряду со многими другими мембраносвязанными пептидами и белками. [53] Недавно Костас Маранас и его коллеги разработали автоматизированный инструмент. [54] изменить размер пор порина внешней мембраны типа F (OmpF) из E.coli до любого желаемого размера субнм и собрать их в мембраны для выполнения точного разделения в ангстремном масштабе.
Другие приложения
[ редактировать ]Одно из наиболее желательных применений дизайна белков — это биосенсоры , белки, которые будут чувствовать присутствие определенных соединений. Некоторые попытки создания биосенсоров включают датчики для неприродных молекул, включая тротил . [55] Совсем недавно Кульман и его коллеги разработали биосенсор PAK1 . [56]
В каком-то смысле белковый дизайн — это разновидность дизайна батарей . [ нужны дальнейшие объяснения ]
См. также
[ редактировать ]- Программное обеспечение для молекулярного дизайна
- Белковая инженерия
- Программное обеспечение для прогнозирования структуры белка
- Сравнение программного обеспечения для моделирования молекулярной механики
Ссылки
[ редактировать ]- ^ Корендович Иван (19 марта 2018 г.). «Минималистический дизайн пептидных и белковых катализаторов» . Американское химическое общество . Проверено 22 марта 2018 г.
- ^ Перейти обратно: а б с д и ж Ричардсон, Дж. С.; Ричардсон, округ Колумбия (июль 1989 г.). «Дизайн белковых структур de novo». Тенденции биохимических наук . 14 (7): 304–9. дои : 10.1016/0968-0004(89)90070-4 . ПМИД 2672455 .
- ^ Перейти обратно: а б с Дахият, Би; Мэйо, SL (3 октября 1997 г.). «Дизайн белка de novo: полностью автоматизированный выбор последовательности». Наука . 278 (5335): 82–7. CiteSeerX 10.1.1.72.7304 . дои : 10.1126/science.278.5335.82 . ПМИД 9311930 .
- ^ Перейти обратно: а б Гордон, Д.Б.; Маршалл, ЮАР; Мэйо, SL (август 1999 г.). «Энергетические функции для дизайна белков». Современное мнение в области структурной биологии . 9 (4): 509–13. дои : 10.1016/s0959-440x(99)80072-4 . ПМИД 10449371 .
- ^ Перейти обратно: а б Харбери, ПБ; Плеч, Джей Джей; Тидор, Б; Альбер, Т; Ким, П.С. (20 ноября 1998 г.). «Дизайн белка высокого разрешения со свободой скелета». Наука . 282 (5393): 1462–7. дои : 10.1126/science.282.5393.1462 . ПМИД 9822371 .
- ^ Перейти обратно: а б с Кульман, Б; Дантас, Г; Иретон, GC; Варани, Г; Стоддард, БЛ; Бейкер, Д. (21 ноября 2003 г.). «Разработка новой складки глобулярного белка с точностью на атомном уровне». Наука . 302 (5649): 1364–8. Бибкод : 2003Sci...302.1364K . дои : 10.1126/science.1089427 . ПМИД 14631033 . S2CID 1939390 .
- ^ Стернер, Р; Меркл, Р; Раушель, FM (май 2008 г.). «Вычислительный дизайн ферментов» . Химия и биология . 15 (5): 421–3. doi : 10.1016/j.chembiol.2008.04.007 . ПМИД 18482694 .
- ^ Ву, Х; Ян, З.Ы.; Ли, Ю; Хогеркорп, CM; Шиф, WR; Моряк, MS; Чжоу, Т; Шмидт, С.Д.; Ву, Л; Сюй, Л; Лонго, Н.С.; Макки, К; О'Делл, С; Громче, МК; Викафф, Д.Л.; Фэн, Ю; Нэйсон, М; Дориа-Роуз, Н.; Коннорс, М; Квонг, PD; Редерер, М; Вятт, RT; Набель, Дж.Дж. ; Маскола-младший (13 августа 2010 г.). «Рациональный дизайн оболочки идентифицирует широко нейтрализующие человеческие моноклональные антитела к ВИЧ-1» . Наука . 329 (5993): 856–61. Бибкод : 2010Sci...329..856W . дои : 10.1126/science.1187659 . ПМК 2965066 . ПМИД 20616233 .
- ^ Хёккер, Б. (8 ноября 2012 г.). «Структурная биология: набор инструментов для дизайна белков» . Природа . 491 (7423): 204–5. Бибкод : 2012Natur.491..204H . дои : 10.1038/491204a . ПМИД 23135466 . S2CID 4426247 .
- ^ Перейти обратно: а б с Ловелл, Южная Каролина; Слово, Дж. М.; Ричардсон, Дж. С.; Ричардсон, округ Колумбия (15 августа 2000 г.). «Предпоследняя библиотека ротамеров». Белки . 40 (3): 389–408. CiteSeerX 10.1.1.555.4071 . doi : 10.1002/1097-0134(20000815)40:3<389::AID-PROT50>3.0.CO;2-2 . ПМИД 10861930 . S2CID 3055173 .
- ^ Шаповалов М.В.; Данбрек Р.Л.-младший (8 июня 2011 г.). «Сглаженная библиотека ротамеров, зависящая от основной цепи, для белков, полученная на основе адаптивных оценок плотности ядра и регрессий» . Структура . 19 (6): 844–58. дои : 10.1016/j.str.2011.03.019 . ПМК 3118414 . ПМИД 21645855 .
- ^ Перейти обратно: а б Самиш, я; МакДермейд, CM; Перес-Агилар, JM; Савен, Дж. Г. (2011). «Теоретический и вычислительный дизайн белка». Ежегодный обзор физической химии . 62 : 129–49. Бибкод : 2011ARPC...62..129S . doi : 10.1146/annurev-physchem-032210-103509 . ПМИД 21128762 .
- ^ Перейти обратно: а б Манделл, диджей; Кортемме, Т. (август 2009 г.). «Гибкость магистрали в вычислительном дизайне белков» (PDF) . Современное мнение в области биотехнологии . 20 (4): 420–8. дои : 10.1016/j.copbio.2009.07.006 . ПМИД 19709874 .
- ^ Перейти обратно: а б с д и ж Дональд, Брюс Р. (2011). Алгоритмы в структурной молекулярной биологии . Кембридж, Массачусетс: MIT Press.
- ^ Перейти обратно: а б Боас, Ф.Е. и Харбери, П.Б. (2007). «Потенциальные энергетические функции для дизайна белков». Современное мнение в области структурной биологии . 17 (2): 199–204. дои : 10.1016/j.sbi.2007.03.006 . ПМИД 17387014 .
- ^ Перейти обратно: а б с д Боас, FE; Харбери, ПБ (апрель 2007 г.). «Потенциальные энергетические функции для дизайна белков». Современное мнение в области структурной биологии . 17 (2): 199–204. дои : 10.1016/j.sbi.2007.03.006 . ПМИД 17387014 .
- ^ Вискарра, CL; Мэйо, SL (декабрь 2005 г.). «Электростатика в вычислительном дизайне белков». Современное мнение в области химической биологии . 9 (6): 622–6. дои : 10.1016/j.cbpa.2005.10.014 . ПМИД 16257567 .
- ^ Чжоу, Т; Георгиев, И; Ву, Х; Ян, З.Ы.; Дай, К; Финци, А; Квон, Ю.Д.; Шайд, Дж. Ф.; Ши, Вт; Сюй, Л; Ян, Ю; Чжу, Дж; Нусенцвейг, MC; Содроски, Дж; Шапиро, Л; Набель, Г.Дж.; Маскола, младший; Квонг, П. Д. (13 августа 2010 г.). «Структурная основа широкой и мощной нейтрализации ВИЧ-1 антителом VRC01» . Наука . 329 (5993): 811–7. Бибкод : 2010Sci...329..811Z . дои : 10.1126/science.1192819 . ПМЦ 2981354 . ПМИД 20616231 .
{{cite journal}}: CS1 maint: несколько имен: список авторов ( ссылка ) - ^ Мендес, Дж; Геруа, Р; Серрано, Л. (август 2002 г.). «Оценка энергии в дизайне белков». Современное мнение в области структурной биологии . 12 (4): 441–6. дои : 10.1016/s0959-440x(02)00345-7 . ПМИД 12163065 .
- ^ Пирс, Северная Каролина; Уинфри, Э. (октябрь 2002 г.). «Дизайн белка NP-труден» . Белковая инженерия . 15 (10): 779–82. дои : 10.1093/протеин/15.10.779 . ПМИД 12468711 .
- ^ Перейти обратно: а б Фойгт, Калифорния; Гордон, Д.Б.; Мэйо, SL (9 июня 2000 г.). «Точность в обмен на скорость: количественное сравнение алгоритмов поиска при проектировании последовательностей белков». Журнал молекулярной биологии . 299 (3): 789–803. CiteSeerX 10.1.1.138.2023 . дои : 10.1006/jmbi.2000.3758 . ПМИД 10835284 .
- ^ Хонг, Э.Дж.; Липпов, С.М.; Тидор, Б; Лосано-Перес, Т. (сентябрь 2009 г.). «Оптимизация ротамера для проектирования белков посредством оценки MAP и уменьшения размера проблемы» . Журнал вычислительной химии . 30 (12): 1923–45. дои : 10.1002/jcc.21188 . ПМК 3495010 . ПМИД 19123203 .
- ^ «Машинное обучение открывает рецепт создания искусственных белков» . физ.орг . Проверено 17 августа 2020 г.
- ^ Расс, Уильям П.; Фиглюцци, Маттео; Стокер, Кристиан; Барра-Шарле, Пьер; Соколич, Михаил; Каст, Питер; Хилверт, Дональд; Монассон, Реми; Кокко, Симона; Вейгт, Мартин; Ранганатан, Рама (2020). «Эволюционная модель создания ферментов хоризматемутазы». Наука . 369 (6502): 440–445. Бибкод : 2020Sci...369..440R . дои : 10.1126/science.aba3304 . ПМИД 32703877 . S2CID 220714458 .
- ^ «Биологи обучают ИИ создавать лекарства и вакцины» . Медицинский центр Вашингтонского университета в Харборвью .
- ^ Ван, Цзюэ; Лисанза, Сидней; Юргенс, Дэвид; Тишер, Дуг; Уотсон, Джозеф Л.; Кастро, Карла М.; Раготт, Роберт; Сарагови, Амиджай; Миллес, Лукас Ф.; Пэк, Минкён; Анищенко Иван; Ян, Вэй; Хикс, Деррик Р.; Экспозит, Марк; Шлихтаерле, Томас; Чун, Юнг-Хо; Даупарас, Юстас; Беннетт, Натаниэль; Вики, Базиль И.М.; Муэнкс, Эндрю; ДиМайо, Фрэнк; Коррейя, Бруно; Овчинников Сергей; Бейкер, Дэвид (22 июля 2022 г.). «Создание функциональных участков белка с использованием глубокого обучения» (PDF) . Наука . 377 (6604): 387–394. Бибкод : 2022Sci...377..387W . дои : 10.1126/science.abn2100 . ISSN 0036-8075 . ПМЦ 9621694 . ПМИД 35862514 .
- ^ Гордон, Д.Б.; Мэйо, SL (15 сентября 1999 г.). «Разветвление и завершение: алгоритм комбинаторной оптимизации дизайна белков» . Структура . 7 (9): 1089–98. дои : 10.1016/s0969-2126(99)80176-2 . ПМИД 10508778 .
- ^ Перейти обратно: а б Лич, Арканзас; Лемон, AP (1 ноября 1998 г.). «Исследование конформационного пространства боковых цепей белка с использованием устранения тупиковых концов и алгоритма A *». Белки . 33 (2): 227–39. CiteSeerX 10.1.1.133.7986 . doi : 10.1002/(sici)1097-0134(19981101)33:2<227::aid-prot7>3.0.co;2-f . ПМИД 9779790 . S2CID 12872539 .
- ^ Перейти обратно: а б Кингсфорд, CL; Шазель, Б; Сингх, М. (1 апреля 2005 г.). «Решение и анализ проблем позиционирования боковой цепи с использованием линейного и целочисленного программирования» . Биоинформатика . 21 (7): 1028–36. doi : 10.1093/биоинформатика/bti144 . ПМИД 15546935 .
- ^ Яновер, Чен; Талия Мельцер; Яир Вайс (2006). «Релаксация линейного программирования и распространение убеждений - эмпирическое исследование». Журнал исследований машинного обучения . 7 : 1887–1907.
- ^ Уэйнрайт, Мартин Дж; Томми С. Яаккола; Алан С. Уиллски (2005). «Оценка MAP посредством соглашения о деревьях: передача сообщений и линейное программирование». Транзакции IEEE по теории информации . 51 (11): 3697–3717. CiteSeerX 10.1.1.71.9565 . дои : 10.1109/тит.2005.856938 . S2CID 10007532 .
- ^ Колмогоров Владимир (28 октября 2006 г.). «Передача сообщений с перевзвешиванием по конвергентному дереву для минимизации энергопотребления». Транзакции IEEE по анализу шаблонов и машинному интеллекту . 28 (10): 1568–1583. дои : 10.1109/TPAMI.2006.200 . ПМИД 16986540 . S2CID 8616813 .
- ^ Глоберсон, Амир; Томми С. Яаккола (2007). «Фиксация максимального продукта: конвергентные алгоритмы передачи сообщений для MAP LP-релаксации». Достижения в области нейронных систем обработки информации .
- ^ Аллен, Б.Д.; Мэйо, SL (30 июля 2006 г.). «Значительное повышение производительности алгоритма оптимизации FASTER». Журнал вычислительной химии . 27 (10): 1071–5. CiteSeerX 10.1.1.425.5418 . дои : 10.1002/jcc.20420 . ПМИД 16685715 . S2CID 769053 .
- ^ Десмет, Дж; Сприт, Дж; Ластерс, I (1 июля 2002 г.). «Быстрая и точная топология боковой цепи и уточнение энергии (FASTER) как новый метод оптимизации структуры белка». Белки . 48 (1): 31–43. дои : 10.1002/прот.10131 . ПМИД 12012335 . S2CID 21524437 .
- ^ Бейкер, Д. (октябрь 2010 г.). «Впереди захватывающий, но трудный путь для разработки вычислительных ферментов» . Белковая наука . 19 (10): 1817–9. дои : 10.1002/pro.481 . ПМЦ 2998717 . ПМИД 20717908 .
- ^ Цзян, Линь; Альтхофф, Эрик А.; Клементе, Фернандо Р.; Дойл, Линдси; Ротлисбергер, Даниэла; Зангеллини, Александр; Галлахер, Жасмин Л.; Беткер, Джейми Л.; Танака, Фуджи (2008). «Вычислительный дизайн ретро-альдольных ферментов De Novo» . Наука . 319 (5868): 1387–91. Бибкод : 2008Sci...319.1387J . дои : 10.1126/science.1152692 . ПМЦ 3431203 . ПМИД 18323453 .
- ^ Ретлисбергер, Даниэла; Херсонский, Ольга; Уоллакотт, Эндрю М.; Цзян, Линь; Динси, Джейсон; Беткер, Джейми; Галлахер, Жасмин Л.; Альтхофф, Эрик А.; Зангеллини, Александр (2008). «Катализаторы удаления Кемпа с помощью компьютерного дизайна ферментов» . Природа . 453 (7192): 190–5. Бибкод : 2008Nature.453..190R . дои : 10.1038/nature06879 . ПМИД 18354394 .
- ^ Сигел, Дж. Б.; Зангеллини, А; Ловик, HM; Поцелуй, Г; Ламберт, Арканзас; Сент-Клер, JL; Галлахер, Дж.Л.; Хилверт, Д; Гелб, Миннесота; Стоддард, БЛ; Хоук, КН; Майкл, FE; Бейкер, Д. (16 июля 2010 г.). «Расчетный дизайн ферментативного катализатора стереоселективной бимолекулярной реакции Дильса-Альдера» . Наука . 329 (5989): 309–13. Бибкод : 2010Sci...329..309S . дои : 10.1126/science.1190239 . ПМК 3241958 . ПМИД 20647463 .
{{cite journal}}: CS1 maint: несколько имен: список авторов ( ссылка ) - ^ Приветт, Гонконг; Поцелуй, Г; Ли, ТМ; Бломберг, Р; Чика, РА; Томас, LM; Хилверт, Д; Хоук, КН; Мэйо, SL (6 марта 2012 г.). «Итеративный подход к компьютерному проектированию ферментов» . Труды Национальной академии наук Соединенных Штатов Америки . 109 (10): 3790–5. Бибкод : 2012PNAS..109.3790P . дои : 10.1073/pnas.1118082108 . ПМК 3309769 . ПМИД 22357762 .
- ^ Чен, CY; Георгиев, И; Андерсон, AC; Дональд, БР (10 марта 2009 г.). «Перестройка активности ферментов на основе вычислительной структуры» . Труды Национальной академии наук Соединенных Штатов Америки . 106 (10): 3764–9. Бибкод : 2009PNAS..106.3764C . дои : 10.1073/pnas.0900266106 . ПМЦ 2645347 . ПМИД 19228942 .
- ^ Корендович, Иван В. (2018). «Рациональный и полурациональный дизайн белка». Белковая инженерия . Методы молекулярной биологии (Клифтон, Нью-Джерси). Том. 1685. стр. 15–23. дои : 10.1007/978-1-4939-7366-8_2 . ISBN 978-1-4939-7364-4 . ISSN 1064-3745 . ПМЦ 5912912 . ПМИД 29086301 .
- ^ Перейти обратно: а б с д Караниколас, Дж; Кульман, Б. (август 2009 г.). «Вычислительный дизайн сродства и специфичности межбелковых интерфейсов» . Современное мнение в области структурной биологии . 19 (4): 458–63. дои : 10.1016/j.sbi.2009.07.005 . ПМК 2882636 . ПМИД 19646858 .
- ^ Шойчет, Б.К. (октябрь 2007 г.). «Нет бесплатного энергетического обеда». Природная биотехнология . 25 (10): 1109–10. дои : 10.1038/nbt1007-1109 . ПМИД 17921992 . S2CID 5527226 .
- ^ Липпов, С.М.; Витруп, К.Д.; Тидор, Б. (октябрь 2007 г.). «Вычислительный дизайн улучшения аффинности антител за пределами созревания in vivo» . Природная биотехнология . 25 (10): 1171–6. дои : 10.1038/nbt1336 . ПМК 2803018 . ПМИД 17891135 .
- ^ Шрайбер, Г; Китинг, А.Е. (февраль 2011 г.). «Специфичность связывания белка и распущенность» . Современное мнение в области структурной биологии . 21 (1): 50–61. дои : 10.1016/j.sbi.2010.10.002 . ПМК 3053118 . ПМИД 21071205 .
- ^ Григорян Г; Рейнке, AW; Китинг, А.Э. (16 апреля 2009 г.). «Определение специфичности взаимодействия с белками дает селективные bZIP-связывающие пептиды» . Природа . 458 (7240): 859–64. Бибкод : 2009Natur.458..859G . дои : 10.1038/nature07885 . ПМЦ 2748673 . ПМИД 19370028 .
- ^ Фрей, К.М.; Георгиев, И; Дональд, БР; Андерсон, AC (3 августа 2010 г.). «Прогнозирование мутаций устойчивости с помощью алгоритмов проектирования белков» . Труды Национальной академии наук Соединенных Штатов Америки . 107 (31): 13707–12. Бибкод : 2010PNAS..10713707F . дои : 10.1073/pnas.1002162107 . ПМЦ 2922245 . ПМИД 20643959 .
- ^ Хури, Джорджия; Фазелиния, Х; Чин, Дж.В.; Пантазес, Р.Дж.; Чирино, ПК; Маранас, CD (октябрь 2009 г.). «Вычислительный дизайн ксилозоредуктазы Candida boidinii для изменения специфичности кофактора» . Белковая наука . 18 (10): 2125–38. дои : 10.1002/pro.227 . ПМЦ 2786976 . ПМИД 19693930 .
- ^ Бертон, ДР; Вайс, РА (13 августа 2010 г.). «СПИД/ВИЧ. Стимул для разработки вакцины против ВИЧ». Наука . 329 (5993): 770–3. Бибкод : 2010Sci...329..770B . дои : 10.1126/science.1194693 . ПМИД 20705840 . S2CID 206528638 .
- ^ Джессика Маршалл (7 ноября 2012 г.). «Белки на заказ» . Новости природы . Проверено 17 ноября 2012 г.
- ^ Разработаны трансмембранные альфа-шпильки в базе данных OPM.
- ^ Разработанные мембраносвязанные пептиды и белки в базе данных OPM.
- ^ Чоудхури, Ратул; Кумар, Маниш; Маранас, Костас Д.; Голбек, Джон Х.; Бейкер, Кэрол; Прабхакар, Дживан; Гривуд, Мэтью; Декер, Карл; Шанкла, Маниш (10 сентября 2018 г.). «PoreDesigner для настройки селективности растворенных веществ в прочных и высокопроницаемых порах внешней мембраны» . Природные коммуникации . 9 (1): 3661. Бибкод : 2018NatCo...9.3661C . дои : 10.1038/s41467-018-06097-1 . ISSN 2041-1723 . ПМК 6131167 . ПМИД 30202038 .
- ^ Лугер, Лорен Л.; Дуайер, Мэри А.; Смит, Джеймс Дж. и Хеллинга, Хомм В. (2003). «Вычислительный дизайн рецепторных и сенсорных белков с новыми функциями». Природа . 423 (6936): 185–190. Бибкод : 2003Natur.423..185L . дои : 10.1038/nature01556 . ПМИД 12736688 . S2CID 4387641 .
- ^ Джа, РК; Ву, Ю.И.; Завистовский, Дж.С.; МакНевин, К; Хан, К.М.; Кульман, Б. (21 октября 2011 г.). «Редизайн автоингибирующего домена PAK1 для повышения стабильности и сродства в биосенсорных приложениях» . Журнал молекулярной биологии . 413 (2): 513–22. дои : 10.1016/j.jmb.2011.08.022 . ПМК 3202338 . ПМИД 21888918 .
Дальнейшее чтение
[ редактировать ]- Дональд, Брюс Р. (2011). Алгоритмы в структурной молекулярной биологии . Кембридж, Массачусетс: MIT Press.
- Сандер, Крис; Фрейнд, Геррит; Базан, Фернандо; Горовиц, Амнон; Накамура, Харуки; Рибас, Луис; Финкельштейн, Алексей В.; Локхарт, Эндрю; Меркл, Райнер; и др. (1992). «Белковый дизайн на компьютерах. Пять новых белков: Шпилька, Грендель, Fingerclasp, Leather и Aida». Белки: структура, функции и биоинформатика . 12 (2): 105–110. дои : 10.1002/прот.340120203 . ПМИД 1603799 . S2CID 38986245 .
- Цзинь, Вэньчжэнь; Камбара, Оки; Сасакава, Хироаки; Тамура, Ацуо и Такада, Сёдзи (2003). «Дизайн складных белков De Novo с гладкой складчатой воронкой: автоматизированный негативный дизайн и экспериментальная проверка» . Структура . 11 (5): 581–590. дои : 10.1016/S0969-2126(03)00075-3 . ПМИД 12737823 .
- Покала, Навин и Гендель, Трейси М. (2005). «Энергетические функции для дизайна белка: корректировка с учетом сродства белок-белковых комплексов, модели развернутого состояния и негативный дизайн растворимости и специфичности». Журнал молекулярной биологии . 347 (1): 203–227. дои : 10.1016/j.jmb.2004.12.019 . ПМИД 15733929 .