восстановление ДНК

Репарация ДНК — это совокупность процессов, с помощью которых клетка идентифицирует и исправляет повреждения молекул ДНК , кодирующих ее геном . [ 1 ] В клетках человека как нормальная метаболическая деятельность, так и факторы окружающей среды, такие как радиация , могут вызвать повреждение ДНК, что приводит к десяткам тысяч отдельных молекулярных повреждений на клетку в день. [ 2 ] Многие из этих повреждений вызывают структурные повреждения молекулы ДНК и могут изменить или устранить способность клетки транскрибировать ген , который кодирует пораженная ДНК. Другие поражения вызывают потенциально опасные мутации в геноме клетки, которые влияют на выживаемость дочерних клеток после митоза . Как следствие, процесс репарации ДНК постоянно активен, поскольку он реагирует на повреждение структуры ДНК. Когда нормальные процессы восстановления терпят неудачу и когда клеточного апоптоза не происходит , может произойти непоправимое повреждение ДНК. В конечном итоге это может привести к злокачественным опухолям или раку согласно гипотезе двух ударов .
Скорость репарации ДНК зависит от различных факторов, включая тип клеток, возраст клетки и внеклеточную среду. Клетка, накопившая большое количество повреждений ДНК или больше не способная эффективно восстанавливать свою ДНК, может войти в одно из трех возможных состояний:
- необратимое состояние покоя, известное как старение
- клеточное самоубийство, также известное как апоптоз или запрограммированная гибель клеток.
- образованию опухоли раковой нерегулируемое деление клеток, что может привести к
Способность клетки восстанавливать ДНК жизненно важна для целостности ее генома и, следовательно, для нормального функционирования организма. Многие гены, влияние которых первоначально было показано на продолжительность жизни, оказались вовлеченными в восстановление и защиту повреждений ДНК. [ 3 ]
2015 года Нобелевская премия по химии была присуждена Томасу Линдалю , Полу Модричу и Азизу Санкару за работу над молекулярными механизмами процессов репарации ДНК. [ 4 ] [ 5 ]
повреждение ДНК
[ редактировать ]Повреждение ДНК, вызванное факторами окружающей среды и нормальными метаболическими процессами внутри клетки, происходит со скоростью от 10 000 до 1 000 000 молекулярных повреждений на клетку в день. [ 2 ] Хотя это составляет не более 0,0003125% от примерно 3,2 миллиардов оснований генома человека, невосстановленные повреждения критических генов (таких как гены-супрессоры опухолей ) могут препятствовать способности клетки выполнять свои функции и значительно увеличивать вероятность образования опухоли и способствовать гетерогенность опухоли .
Подавляющее большинство повреждений ДНК затрагивает первичную структуру двойной спирали; то есть сами основания химически модифицированы. Эти модификации, в свою очередь, могут нарушить регулярную спиральную структуру молекул за счет введения неродных химических связей или объемных аддуктов , которые не вписываются в стандартную двойную спираль. В отличие от белков и РНК , ДНК обычно не имеет третичной структуры , поэтому на этом уровне не происходит повреждений или нарушений. Однако ДНК сверхспиральна и намотана вокруг «упаковочных» белков, называемых гистонами (у эукариот), и обе суперструктуры уязвимы к последствиям повреждения ДНК.
Источники
[ редактировать ]Повреждения ДНК можно разделить на два основных типа:
- эндогенные повреждения, такие как атака активных форм кислорода, образующихся из побочных продуктов нормального метаболизма (спонтанная мутация), особенно процесс окислительного дезаминирования
- также включает ошибки репликации
- экзогенное повреждение, вызванное внешними агентами, такими как
- ультрафиолетовое (УФ) излучение (200–400 нм ) солнца или других искусственных источников света
- другие частоты излучения, включая рентгеновские лучи и гамма-лучи
- гидролиз или термическое разрушение
- некоторые растительные токсины
- , созданные человеком мутагенные химические вещества , особенно ароматические соединения, которые действуют как интеркалирующие агенты ДНК.
- вирусы [ 6 ]
Репликация поврежденной ДНК до деления клетки может привести к включению неправильных оснований напротив поврежденных. Дочерние клетки, наследующие эти неправильные основания, несут мутации, из-за которых исходную последовательность ДНК невозможно восстановить (за исключением редких случаев обратной мутации , например, посредством конверсии генов ).
Типы
[ редактировать ]Существует несколько типов повреждений ДНК, обусловленных эндогенными клеточными процессами:
- окисление оснований [например, 8-оксо-7,8-дигидрогуанина (8-oxoG)] и образование разрывов цепи ДНК из активных форм кислорода,
- алкилирование оснований (обычно метилирование ), такое как образование 7-метилгуанозина , 1-метиладенина, 6-О-метилгуанина
- гидролиз оснований, такой как дезаминирование , депуринизация и депиримидинирование.
- «объемное образование аддукта» (например, аддукт бензо[а]пирендиол-эпоксид-dG, аддукт аристолактама I-dA)
- несовпадение оснований из-за ошибок в репликации ДНК , при которых неправильное основание ДНК пришивается на место во вновь образующейся цепи ДНК или основание ДНК пропускается или вставляется по ошибке.
- Повреждение моноаддукта, вызванное изменением одного азотистого основания ДНК
- При повреждении аддукта
Повреждения, вызванные экзогенными агентами, проявляются во многих формах. Некоторые примеры:
- УФ-В-свет вызывает сшивание между соседними основаниями цитозина и тимина , образуя димеры пиримидина . Это называется прямым повреждением ДНК .
- УФ-А свет создает в основном свободные радикалы . Повреждение, вызванное свободными радикалами, называется непрямым повреждением ДНК .
- Ионизирующее излучение , например, создаваемое радиоактивным распадом или космическими лучами, вызывает разрывы нитей ДНК. Ионизирующее излучение среднего уровня может вызвать непоправимое повреждение ДНК (приводящее к ошибкам репликации и транскрипции, необходимым для неоплазии, или может вызвать вирусные взаимодействия), что приводит к преждевременному старению и раку.
- Термическое разрушение при повышенной температуре увеличивает скорость депуринации (потеря пуриновых оснований из основной цепи ДНК) и одноцепочечных разрывов. Например, гидролитическое депуринирование наблюдается у термофильных бактерий , которые растут в горячих источниках при температуре 40–80 °C. [ 7 ] [ 8 ] Скорость депуринации (300 пуриновых возможность адаптивного ответа . остатков на геном на поколение) у этих видов слишком высока, чтобы ее можно было восстановить с помощью обычных механизмов репарации, поэтому нельзя исключать
- Промышленные химикаты, такие как винилхлорид и перекись водорода , а также химикаты окружающей среды, такие как полициклические ароматические углеводороды, содержащиеся в дыме, саже и смоле, создают огромное разнообразие аддуктов ДНК — этаноатов, окисленных оснований, алкилированных фосфодиэфиров и сшивок ДНК , и это лишь некоторые из них. .
УФ-повреждение, алкилирование/метилирование, рентгеновское повреждение и окислительное повреждение являются примерами индуцированного повреждения. Спонтанное повреждение может включать потерю основания, дезаминирование, сморщивание сахарного кольца и таутомерный сдвиг. Конститутивные (спонтанные) повреждения ДНК, вызванные эндогенными оксидантами, можно обнаружить по низкому уровню фосфорилирования гистона H2AX в необработанных клетках. [ 9 ]
Ядерный против митохондриального
[ редактировать ]В клетках человека и эукариотических клетках в целом ДНК находится в двух клеточных местах – внутри ядра и внутри митохондрий . Ядерная ДНК (н-ДНК) существует в виде хроматина на нерепликативных стадиях клеточного цикла и конденсируется в агрегатные структуры, известные как хромосомы, во время деления клеток . В любом из этих состояний ДНК сильно уплотнена и свернута вокруг белков, похожих на шарики, называемых гистонами . Всякий раз, когда клетке необходимо выразить генетическую информацию, закодированную в ее н-ДНК, требуемая хромосомная область раскрывается, расположенные в ней гены экспрессируются, а затем область конденсируется обратно в свою конформацию покоя. Митохондриальная ДНК (мтДНК) расположена внутри митохондриальных органелл , существует в нескольких копиях, а также тесно связана с рядом белков, образуя комплекс, известный как нуклеоид. Внутри митохондрий активные формы кислорода (АФК) или свободные радикалы , побочные продукты постоянного производства аденозинтрифосфата (АТФ) посредством окислительного фосфорилирования. , создают высокоокислительную среду, которая, как известно, повреждает мтДНК. Важнейшим ферментом в противодействии токсичности этих видов является супероксиддисмутаза , которая присутствует как в митохондриях, так и в цитоплазме эукариотических клеток.
Старение и апоптоз
[ редактировать ]Старение, необратимый процесс, при котором клетка больше не делится , является защитной реакцией на укорочение концов хромосом, называемых теломерами . Теломеры представляют собой длинные участки повторяющейся некодирующей ДНК , которые покрывают хромосомы и подвергаются частичной деградации каждый раз, когда клетка подвергается делению (см. предел Хейфлика ). [ 10 ] Напротив, покой — это обратимое состояние клеточного покоя, не связанное с повреждением генома (см. клеточный цикл ). Старение клеток может служить функциональной альтернативой апоптозу в тех случаях, когда физическое присутствие клетки по пространственным причинам требуется организму. [ 11 ] который служит механизмом «последнего средства», предотвращающим ненадлежащую репликацию клетки с поврежденной ДНК в отсутствие передачи сигналов, способствующих росту клеток . Нерегулируемое деление клеток может привести к образованию опухоли (см. рак ), потенциально смертельной для организма. Поэтому индукция старения и апоптоза считается частью стратегии защиты от рака. [ 12 ]
Мутация
[ редактировать ]Важно различать повреждение ДНК и мутацию — два основных типа ошибок в ДНК. Повреждение ДНК и мутация принципиально различны. Повреждения приводят к физическим аномалиям в ДНК, таким как одно- и двухцепочечные разрывы, остатки 8-гидроксидезоксигуанозина и аддукты полициклических ароматических углеводородов. Повреждения ДНК могут быть распознаны ферментами и, таким образом, могут быть правильно восстановлены, если для копирования доступна избыточная информация, такая как неповрежденная последовательность в комплементарной цепи ДНК или в гомологичной хромосоме. Если в клетке сохраняется повреждение ДНК, транскрипция гена может быть предотвращена, и, таким образом, трансляция в белок также будет заблокирована. Репликация также может быть заблокирована или клетка может погибнуть.
В отличие от повреждения ДНК, мутация представляет собой изменение базовой последовательности ДНК. Мутация не может быть распознана ферментами, если изменение оснований присутствует в обеих цепях ДНК, и, следовательно, мутация не может быть исправлена. На клеточном уровне мутации могут вызывать изменения в функции и регуляции белков. Мутации реплицируются при репликации клетки. В популяции клеток частота появления мутантных клеток будет увеличиваться или уменьшаться в зависимости от влияния мутации на способность клетки выживать и размножаться.
Хотя повреждения ДНК и мутации явно отличаются друг от друга, они связаны между собой, поскольку повреждение ДНК часто вызывает ошибки синтеза ДНК во время репликации или репарации; эти ошибки являются основным источником мутаций.
Учитывая эти свойства повреждений и мутаций ДНК, можно видеть, что повреждение ДНК представляет собой особую проблему в неделящихся или медленно делящихся клетках, где неустраненные повреждения имеют тенденцию накапливаться с течением времени. С другой стороны, в быстро делящихся клетках невосстановленные повреждения ДНК, которые не убивают клетку путем блокирования репликации, будут иметь тенденцию вызывать ошибки репликации и, следовательно, мутацию. Подавляющее большинство мутаций, которые не являются нейтральными по своему эффекту, вредны для выживания клетки. Таким образом, в популяции клеток, составляющей ткань с реплицирующими клетками, мутантные клетки имеют тенденцию к потере. Однако нечастые мутации, обеспечивающие преимущество в выживании, будут иметь тенденцию к клональной экспансии за счет соседних клеток в ткани. Это преимущество для клетки невыгодно для всего организма, поскольку такие мутантные клетки могут вызывать рак. Таким образом, повреждение ДНК в часто делящихся клетках, поскольку оно приводит к мутациям, является основной причиной рака. Напротив, повреждение ДНК в редко делящихся клетках, вероятно, является основной причиной старения. [ 13 ]
Механизмы
[ редактировать ]Клетки не могут функционировать, если повреждение ДНК нарушает целостность и доступность важной информации в геноме (но клетки остаются поверхностно функциональными, когда несущественные гены отсутствуют или повреждены). В зависимости от типа повреждения двойной спиральной структуры ДНК возникли различные стратегии восстановления потерянной информации. Если возможно, клетки используют немодифицированную комплементарную цепь ДНК или сестринскую хроматиду в качестве матрицы для восстановления исходной информации. Без доступа к шаблону клетки используют подверженный ошибкам механизм восстановления, известный как трансочаговый синтез в крайнем случае .
Повреждение ДНК изменяет пространственную конфигурацию спирали, и такие изменения могут быть обнаружены клеткой. Как только повреждение локализовано, определенные молекулы восстановления ДНК связываются в месте повреждения или рядом с ним, побуждая другие молекулы связываться и образовывать комплекс, который позволяет осуществить фактическое восстановление.
Прямой разворот
[ редактировать ]Известно, что клетки устраняют три типа повреждений своей ДНК, химически обращая их. Эти механизмы не требуют шаблона, поскольку типы повреждений, которым они противодействуют, могут возникать только на одной из четырех баз. Такие механизмы прямого обращения специфичны для типа нанесенного повреждения и не связаны с разрушением фосфодиэфирного остова. Образование димеров пиримидина при облучении УФ-светом приводит к образованию аномальной ковалентной связи между соседними пиримидиновыми основаниями. Процесс фотореактивации напрямую устраняет это повреждение под действием фермента фотолиазы , активация которого обязательно зависит от энергии, поглощаемой синим/УФ-светом 300–500 нм ( длина волны ) для стимулирования катализа. [ 14 ] Фотолиаза, старый фермент, присутствующий в бактериях , грибах и большинстве животных , больше не функционирует в организме человека. [ 15 ] которые вместо этого используют эксцизионную репарацию нуклеотидов для устранения повреждений, вызванных УФ-облучением. Другой тип повреждения, метилирование оснований гуанина, напрямую устраняется ферментом метилгуанинметилтрансферазой (MGMT), бактериальный эквивалент которого называется ogt . Это дорогостоящий процесс, поскольку каждую молекулу MGMT можно использовать только один раз; то есть реакция является стехиометрической, а не каталитической . [ 16 ] Генерализованный ответ бактерий на метилирующие агенты известен как адаптивный ответ и обеспечивает определенный уровень устойчивости к алкилирующим агентам при длительном воздействии за счет повышения регуляции ферментов восстановления алкилирования. [ 17 ] Третий тип повреждения ДНК, обратимый клетками, — это определенное метилирование оснований цитозина и аденина.
Однонитевое повреждение
[ редактировать ]
Когда только одна из двух нитей двойной спирали имеет дефект, другую нить можно использовать в качестве шаблона для коррекции поврежденной нити. Чтобы восстановить повреждение одной из двух парных молекул ДНК, существует ряд механизмов эксцизионной репарации , которые удаляют поврежденный нуклеотид и заменяют его неповрежденным нуклеотидом, комплементарным нуклеотиду, обнаруженному в неповрежденной цепи ДНК. [ 16 ]
- Эксцизионное восстановление оснований (BER): поврежденные отдельные основания или нуклеотиды чаще всего восстанавливаются путем удаления задействованного основания или нуклеотида и последующей вставки правильного основания или нуклеотида. При базовой эксцизионной репарации гликозилаза [ 18 ] Фермент удаляет поврежденное основание из ДНК, разрывая связь между основанием и дезоксирибозой. Эти ферменты удаляют одно основание, создавая апуриновый или апиримидиновый сайт ( AP-сайт ). [ 18 ] Ферменты, называемые AP-эндонуклеазами, разрывают поврежденный остов ДНК в AP-сайте. Затем ДНК-полимераза удаляет поврежденную область, используя свою экзонуклеазную активность от 5' до 3', и правильно синтезирует новую цепь, используя комплементарную цепь в качестве матрицы. [ 18 ] Затем разрыв закрывается ферментом ДНК-лигазой. [ 19 ]
- Эксцизионная репарация нуклеотидов (NER): объемные повреждения, искажающие спираль, такие как димеризация пиримидина, вызванная УФ-светом, обычно восстанавливаются с помощью трехэтапного процесса. удаляются цепи ДНК длиной 12–24 нуклеотида как выше, так и ниже места повреждения Сначала распознается повреждение, затем эндонуклеазами , а затем удаленный участок ДНК повторно синтезируется. [ 20 ] NER представляет собой высокоэволюционно консервативный механизм восстановления и используется почти во всех эукариотических и прокариотических клетках. [ 20 ] У прокариот NER опосредуется белками Uvr . [ 20 ] У эукариот задействовано гораздо больше белков, хотя общая стратегия та же. [ 20 ]
- Системы исправления несоответствий присутствуют практически во всех ячейках для исправления ошибок, которые не исправляются корректурой . Эти системы состоят как минимум из двух белков. Один обнаруживает несоответствие, а другой привлекает эндонуклеазу, которая расщепляет вновь синтезированную цепь ДНК вблизи области повреждения. В E. coli задействованные белки представляют собой белки класса Mut: MutS, MutL и MutH. У большинства эукариот аналогом MutS является MSH, а аналогом MutL — MLH. MutH присутствует только у бактерий. За этим следует удаление поврежденной области экзонуклеазой, ресинтез ДНК-полимеразой и запечатывание разрывов ДНК-лигазой. [ 21 ]
Двухцепочечные разрывы
[ редактировать ]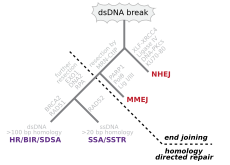
Двухцепочечные разрывы, при которых обе цепи двойной спирали разрываются, особенно опасны для клетки, поскольку могут привести к перестройкам генома . Фактически, когда двухцепочечный разрыв сопровождается поперечной связью, соединяющей две цепи в одной и той же точке, ни одна цепь не может использоваться в качестве матрицы для механизмов репарации, так что клетка не сможет завершить митоз, когда Затем он делится и либо умирает, либо, в редких случаях, подвергается мутации. [ 22 ] [ 23 ] Существуют три механизма восстановления двухцепочечных разрывов (DSB): негомологическое соединение концов (NHEJ), соединение концов, опосредованное микрогомологией (MMEJ), и гомологичная рекомбинация (HR): [ 16 ] [ 24 ]

- В NHEJ ДНК-лигаза IV , специализированная ДНК-лигаза , образующая комплекс с кофактором XRCC4 , непосредственно соединяет два конца. [ 25 ] Чтобы обеспечить точную репарацию, NHEJ полагается на короткие гомологичные последовательности, называемые микрогомологиями, присутствующие на одноцепочечных хвостах соединяемых концов ДНК. Если эти выступы совместимы, ремонт обычно получается точным. [ 26 ] [ 27 ] [ 28 ] [ 29 ] NHEJ также может вносить мутации во время восстановления. Потеря поврежденных нуклеотидов в месте разрыва может привести к делециям, а соединение несовпадающих концов образует инсерции или транслокации. NHEJ особенно важен до того, как клетка реплицирует свою ДНК, поскольку не существует матрицы, доступной для репарации путем гомологичной рекомбинации. существуют «резервные» пути NHEJ У высших эукариот . [ 30 ] Помимо своей роли хранителя генома, NHEJ необходим для соединения двухцепочечных разрывов со шпильками, индуцированных во время рекомбинации V (D) J , процесса, который генерирует разнообразие рецепторов B-клеток и T-клеток в позвоночных иммунной системе . [ 31 ]
- короткого расстояния MMEJ начинается с резекции концов нуклеазой MRE11 по обе стороны от двухцепочечного разрыва, чтобы выявить области микрогомологии. [ 32 ] В дальнейших шагах [ 33 ] Поли(ADP-рибоза)полимераза 1 (PARP1) необходима и может быть ранним шагом в MMEJ. Происходит спаривание областей микрогомологии с последующим привлечением эндонуклеазы 1, специфичной для структуры лоскута (FEN1), для удаления нависающих лоскутов. За этим следует рекрутирование XRCC1 – LIG3 в сайт лигирования концов ДНК, что приводит к образованию интактной ДНК. MMEJ всегда сопровождается делецией, так что MMEJ представляет собой мутагенный путь репарации ДНК. [ 34 ]
- HR требует наличия идентичной или почти идентичной последовательности, которая будет использоваться в качестве матрицы для восстановления разрыва. Ферментативный механизм, ответственный за этот процесс восстановления, почти идентичен механизму, ответственному за хромосомный кроссинговер во время мейоза. Этот путь позволяет восстановить поврежденную хромосому с использованием сестринской хроматиды (доступной в G2 после репликации ДНК) или гомологичной хромосомы в качестве матрицы. DSB, вызванные попытками репликационного аппарата синтезировать одноцепочечный разрыв или невосстановленное повреждение, вызывают коллапс репликационной вилки и обычно восстанавливаются путем рекомбинации.
В системе in vitro MMEJ возникал в клетках млекопитающих на уровне 10–20% от HR, когда также были доступны механизмы HR и NHEJ. [ 32 ]
Экстремофил ионизирующим Deinococcus radiodurans обладает замечательной способностью выдерживать повреждения ДНК, вызванные излучением и другими источниками. По крайней мере две копии генома со случайными разрывами ДНК могут образовывать фрагменты ДНК посредством отжига . Частично перекрывающиеся фрагменты затем используются для синтеза гомологичных областей посредством движущейся D-петли , которая может продолжать удлинение до тех пор, пока не будут найдены комплементарные партнерские цепи. На заключительном этапе происходит кроссинговер посредством RecA -зависимой гомологичной рекомбинации . [ 35 ]
Топоизомеразы вносят как одно-, так и двухцепочечные разрывы в ходе изменения состояния суперспирализации ДНК , что особенно часто встречается в областях вблизи открытой репликационной вилки. Такие разрывы не считаются повреждением ДНК, поскольку они являются естественным промежуточным продуктом биохимического механизма топоизомеразы и немедленно восстанавливаются создавшими их ферментами.
Другой тип двухцепочечных разрывов ДНК происходит из термочувствительных или термолабильных участков ДНК. Эти сайты ДНК не являются исходными DSB. Однако после обработки повышенной температурой они превращаются в ДСБ. Ионизирующее облучение может вызвать очень сложную форму повреждения ДНК в виде кластерного повреждения. Он состоит из различных типов повреждений ДНК в различных местах спирали ДНК. Некоторые из этих близко расположенных поражений, вероятно, могут превратиться в DSB под воздействием высоких температур. Но точная природа этих поражений и их взаимодействие пока не известны. [ 36 ]
Синтез транслезий
[ редактировать ]Синтез транслезий (TLS) — это процесс толерантности к повреждениям ДНК, который позволяет механизму репликации ДНК реплицировать прошлые повреждения ДНК, такие как димеры тимина или AP-сайты . [ 37 ] Он включает в себя замену обычных ДНК-полимераз специализированными полимеразами трансповреждения (т.е. ДНК-полимеразой IV или V из семейства Y-полимераз), часто с более крупными активными сайтами, которые могут облегчить вставку оснований напротив поврежденных нуклеотидов. Считается, что переключение полимеразы опосредовано, среди других факторов, посттрансляционной модификацией фактора репликации процессивности PCNA . Полимеразы транслесионного синтеза часто имеют низкую точность (высокую склонность к вставке неправильных оснований) на неповрежденных матрицах по сравнению с обычными полимеразами. Тем не менее, многие из них чрезвычайно эффективны при вставке правильных баз против конкретных типов повреждений. Например, Pol η обеспечивает безошибочное обход поражений, вызванных УФ-облучением , тогда как Pol ι вызывает мутации в этих сайтах. Известно, что Pol η добавляет первый аденин к фотодимеру T^T с использованием пары оснований Уотсона-Крика , а второй аденин будет добавлен в его син-конформации с использованием пары оснований Хугстина . С точки зрения сотовой связи риск введения точечные мутации во время синтеза транслейкоза могут быть предпочтительнее, чем прибегать к более радикальным механизмам репарации ДНК, которые могут вызвать грубые хромосомные аберрации или гибель клеток. Короче говоря, в этом процессе участвуют специализированные полимеразы, которые либо обходят, либо восстанавливают повреждения в местах остановки репликации ДНК. Например, ДНК-полимераза человека эта может обходить сложные повреждения ДНК, такие как внутрицепочечная сшивка гуанин-тимина, G[8,5-Me]T, хотя она может вызывать целевые и полуцелевые мутации. [ 38 ] Паромита Райчаудхури и Ашис Басу [ 39 ] изучали токсичность и мутагенез того же поражения в Escherichia coli путем репликации G[8,5-Me]T-модифицированной плазмиды в E. coli со специфическими нокаутами ДНК-полимеразы. Жизнеспособность была очень низкой у штамма, лишенного pol II, pol IV и pol V, трех SOS-индуцируемых ДНК-полимераз, что указывает на то, что синтез транслейкоза осуществляется в основном этими специализированными ДНК-полимеразами. Обходную платформу для этих полимераз обеспечивает ядерный антиген пролиферирующих клеток (PCNA). В нормальных условиях PCNA, связанная с полимеразами, реплицирует ДНК. В месте повреждения PCNA убиквитинируется или модифицируется белками RAD6/ , RAD18 обеспечивая платформу для специализированных полимераз, позволяющих обойти повреждение и возобновить репликацию ДНК. [ 40 ] [ 41 ] После синтеза транслезии требуется расширение. Это расширение может быть осуществлено с помощью репликативной полимеразы, если TLS не содержит ошибок, как в случае с Pol η, но если TLS приводит к несоответствию, для его расширения необходима специализированная полимераза; Пол ζ . Pol ζ уникален тем, что может расширять терминальные несоответствия, тогда как более процессивные полимеразы не могут этого сделать. Таким образом, при обнаружении повреждения репликационная вилка останавливается, PCNA переключается с процессивной полимеразы на полимеразу TLS, такую как Pol ι, чтобы исправить повреждение, затем PCNA может переключиться на Pol ζ, чтобы расширить несоответствие, и последняя PCNA переключится. процессивной полимеразе для продолжения репликации.
Глобальный ответ на повреждение ДНК
[ редактировать ]Клетки, подвергшиеся воздействию ионизирующего излучения , ультрафиолетового света или химических веществ, склонны к образованию множественных участков объемных повреждений ДНК и двухцепочечных разрывов. Более того, агенты, повреждающие ДНК, могут повредить другие биомолекулы, такие как белки , углеводы , липиды и РНК . Накопление повреждений, а именно двухцепочечные разрывы или аддукты, останавливающие репликационные вилки , относятся к числу известных сигналов стимуляции глобального ответа на повреждение ДНК. [ 42 ] Глобальный ответ на повреждение представляет собой действие, направленное на сохранение клеток и запускающее множество путей восстановления макромолекул, обхода повреждений, толерантности или апоптоза . Общими чертами глобального ответа являются индукция множества генов , остановка клеточного цикла и ингибирование клеточного деления .
Начальные шаги
[ редактировать ]Упаковка эукариотической ДНК в хроматин представляет собой барьер для всех процессов, основанных на ДНК, которые требуют привлечения ферментов к местам их действия. Чтобы обеспечить восстановление ДНК, хроматин должен быть ремоделирован . У эукариот АТФ- зависимые ремоделирования хроматина комплексы и ферменты, модифицирующие гистоны, являются двумя преобладающими факторами, используемыми для осуществления этого процесса ремоделирования. [ 43 ]
Релаксация хроматина быстро происходит в месте повреждения ДНК. [ 44 ] [ 45 ] На одном из самых ранних этапов активируемая стрессом протеинкиназа, N-концевая киназа c-Jun (JNK) , фосфорилирует SIRT6 по серину 10 в ответ на двухцепочечные разрывы или другие повреждения ДНК. [ 46 ] Эта посттрансляционная модификация облегчает мобилизацию SIRT6 к сайтам повреждения ДНК и необходима для эффективного рекрутирования поли(ADP-рибозо)полимеразы 1 (PARP1) к сайтам разрыва ДНК и для эффективного восстановления DSB. [ 46 ] Белок PARP1 начинает появляться в местах повреждения ДНК менее чем за секунду, с полумаксимальным накоплением в течение 1,6 секунды после возникновения повреждения. [ 47 ] PARP1 синтезирует полимерные цепи аденозиндифосфатрибозы на себе (поли(АДФ-рибоза) или PAR). Затем ремодератор хроматина ALC1 быстро присоединяется к продукту действия PARP1, рибозной цепи поли-АДФ, и ALC1 завершает прибытие к повреждению ДНК в течение 10 секунд после возникновения повреждения. [ 45 ] Около половины максимального расслабления хроматина, предположительно за счет действия ALC1, происходит к 10 с. [ 45 ] Затем это позволяет задействовать фермент репарации ДНК MRE11 , чтобы инициировать репарацию ДНК в течение 13 секунд. [ 47 ]
γH2AX, фосфорилированная форма H2AX, также участвует в ранних стадиях, приводящих к деконденсации хроматина после двухцепочечных разрывов ДНК. Вариант гистонов H2AX составляет около 10% гистонов H2A в хроматине человека. [ 48 ] γH2AX (H2AX, фосфорилированный по серину 139) можно обнаружить уже через 20 секунд после облучения клеток (с образованием двухцепочечного разрыва ДНК), а накопление γH2AX на половину максимального уровня происходит за одну минуту. [ 48 ] Протяженность хроматина с фосфорилированным γH2AX составляет около двух миллионов пар оснований в месте двухцепочечного разрыва ДНК. [ 48 ] γH2AX сам по себе не вызывает деконденсацию хроматина, но в течение 30 секунд после облучения RNF8 в ассоциации с γH2AX. можно обнаружить белок [ 49 ] RNF8 опосредует обширную деконденсацию хроматина посредством его последующего взаимодействия с CHD4 . [ 50 ] компонент комплекса ремоделирования нуклеосом и деацетилазы NuRD .
DDB2 встречается в гетеродимерном комплексе с DDB1 . Этот комплекс далее образует комплексы с убиквитинлигазы белком CUL4A. [ 51 ] и с PARP1 . [ 52 ] Этот более крупный комплекс быстро связывается с повреждением хроматина, вызванным УФ-излучением, при этом ассоциация на половине максимума завершается за 40 секунд. [ 51 ] Белок PARP1, прикрепленный как к DDB1, так и к DDB2, затем PARилирует (создает рибозную цепь поли-АДФ) на DDB2, что привлекает белок ремоделирования ДНК ALC1 . [ 52 ] Действие ALC1 расслабляет хроматин в месте повреждения ДНК УФ-излучением. Это расслабление позволяет другим белкам пути эксцизионной репарации нуклеотидов проникать в хроматин и восстанавливать вызванные УФ-излучением повреждения димера циклобутан-пиримидина .
После быстрого ремоделирования хроматина активируются клеточного цикла точки контрольные , позволяющие восстановить ДНК до того, как клеточный цикл продолжится. Во-первых, две киназы , ATM и ATR, активируются в течение 5 или 6 минут после повреждения ДНК. За этим следует фосфорилирование белка контрольной точки клеточного цикла Chk1 , запускающего его функцию, примерно через 10 минут после повреждения ДНК. [ 53 ]
Контрольные точки повреждения ДНК
[ редактировать ]После повреждения ДНК клеточного цикла контрольные точки активируются . Активация контрольной точки приостанавливает клеточный цикл и дает клетке время восстановить повреждения, прежде чем продолжить деление. Контрольные точки повреждения ДНК возникают на границах G1 / S и G2 / M . внутри S. Также существует контрольная точка Активация контрольной точки контролируется двумя мастер -киназами , ATM и ATR . АТМ реагирует на двухцепочечные разрывы ДНК и нарушения структуры хроматина, [ 54 ] тогда как ATR в первую очередь реагирует на остановку вилок репликации . Эти киназы фосфорилируют нижестоящие мишени в каскаде сигнальной трансдукции , что в конечном итоге приводит к остановке клеточного цикла. класс белков-медиаторов контрольных точек, включая BRCA1 , MDC1 и 53BP1 . Также был идентифицирован [ 55 ] Эти белки, по-видимому, необходимы для передачи сигнала активации контрольной точки нижестоящим белкам.
Контрольная точка повреждения ДНК — это путь передачи сигнала , который блокирует развитие клеточного цикла в G1, G2 и метафазе и замедляет скорость прогрессирования S-фазы при ДНК повреждении . Это приводит к паузе в клеточном цикле, давая клеткам время восстановить повреждения, прежде чем продолжить деление.
Белки контрольной точки можно разделить на четыре группы: фосфатидилинозитол-3-киназа (PI3K)-подобная протеинкиназа , группа, подобная ядерному антигену пролиферирующих клеток (PCNA), две серин/треонин (S/T) киназы и их адаптеры. Центральное место во всех реакциях контрольных точек, индуцированных повреждением ДНК, занимает пара крупных протеинкиназ, принадлежащих к первой группе PI3K-подобных протеинкиназ - киназы ATM ( мутированная атаксия телеангиэктазии ) и ATR (атаксия- и Rad-связанная) киназы, последовательность и функции которых хорошо сохранились в эволюции. Любой ответ на повреждение ДНК требует либо ATM, либо ATR, поскольку они обладают способностью связываться с хромосомами в месте повреждения ДНК вместе с дополнительными белками, которые являются платформами, на которых могут быть собраны компоненты ответа на повреждение ДНК и комплексы репарации ДНК.
Важной последующей мишенью ATM и ATR является р53 , поскольку он необходим для индукции апоптоза после повреждения ДНК. [ 56 ] Ингибитор циклин-зависимой киназы p21 индуцируется как p53-зависимым, так и p53-независимым механизмом и может останавливать клеточный цикл в контрольных точках G1/S и G2/M путем деактивации комплексов циклин / циклин-зависимая киназа . [ 57 ]
Прокариотический SOS-ответ
[ редактировать ]SOS -ответ — это изменения экспрессии генов у Escherichia coli и других бактерий в ответ на обширное повреждение ДНК. SOS- система прокариот регулируется двумя ключевыми белками: LexA и RecA . LexA Годимер представляет собой транскрипции репрессор , который связывается с операторными последовательностями, обычно называемыми SOS-боксами. у Escherichia coli LexA регулирует транскрипцию примерно 48 генов, включая гены lexA и RecA. Известно, что [ 58 ] Известно, что SOS-ответ широко распространен в домене бактерий, но он в основном отсутствует у некоторых типов бактерий, таких как спирохеты . [ 59 ] Наиболее распространенными клеточными сигналами, активирующими SOS-ответ, являются участки одноцепочечной ДНК (оцДНК), возникающие в результате остановки вилок репликации или двухцепочечных разрывов, которые обрабатываются ДНК-хеликазой для разделения двух цепей ДНК. [ 42 ] На этапе инициации белок RecA связывается с оцДНК в реакции, управляемой гидролизом АТФ, создавая нити RecA-оцДНК. Нити RecA-оцДНК активируют активность аутопротеазы LexA , что в конечном итоге приводит к расщеплению димера LexA и последующей деградации LexA. Утрата репрессора LexA индуцирует транскрипцию генов SOS и обеспечивает дальнейшую индукцию сигнала, ингибирование деления клеток и увеличение уровней белков, ответственных за обработку повреждений.
У Escherichia coli SOS-боксы представляют собой последовательности длиной 20 нуклеотидов вблизи промоторов с палиндромной структурой и высокой степенью консервативности последовательности. В других классах и типах последовательность SOS-боксов значительно варьирует, имеет разную длину и состав, но всегда высококонсервативна и является одним из самых сильных коротких сигналов в геноме. [ 59 ] Высокая информативность SOS-боксов позволяет дифференциально связывать LexA с разными промоторами и определять время SOS-ответа. Гены восстановления повреждений индуцируются в начале SOS-ответа. Подверженные ошибкам полимеразы трансфекции, например, UmuCD'2 (также называемая ДНК-полимеразой V), индуцируются позже, в крайнем случае. [ 60 ] После того, как повреждение ДНК восстанавливается или обходится с помощью полимераз или посредством рекомбинации, количество одноцепочечной ДНК в клетках уменьшается, уменьшение количества нитей RecA снижает активность расщепления гомодимера LexA, который затем связывается с SOS-боксами вблизи промоторов и восстанавливает нормальная экспрессия генов.
Транскрипционные реакции эукариот на повреждение ДНК
[ редактировать ]Эукариотические клетки, подвергшиеся воздействию агентов, повреждающих ДНК, также активируют важные защитные пути, индуцируя множество белков, участвующих в репарации ДНК, контроле контрольных точек клеточного цикла , транспортировке и деградации белков. Такой полногеномный транскрипционный ответ очень сложен и жестко регулируется, что позволяет скоординировать глобальный ответ на повреждение. Воздействие на дрожжи Saccharomyces cerevisiae агентов, повреждающих ДНК, приводит к перекрывающимся, но различным профилям транскрипции. Сходство с реакцией на экологический шок указывает на то, что общий глобальный путь реакции на стресс существует на уровне активации транскрипции. Напротив, разные типы клеток человека по-разному реагируют на повреждения, что указывает на отсутствие общего глобального ответа. Вероятное объяснение этой разницы между клетками дрожжей и человека может заключаться в гетерогенности клеток млекопитающих . У животного разные типы клеток распределены по разным органам, которые развили разную чувствительность к повреждению ДНК. [ 61 ]
В целом глобальный ответ на повреждение ДНК включает в себя экспрессию множества генов, ответственных за пострепликационное восстановление , гомологичную рекомбинацию, эксцизионную репарацию нуклеотидов, контрольную точку повреждения ДНК , глобальную активацию транскрипции, гены, контролирующие распад мРНК, и многие другие. Большой объем повреждений клетки ставит ее перед важным решением: подвергнуться апоптозу и умереть или выжить ценой жизни с измененным геномом. Увеличение толерантности к повреждениям может привести к увеличению выживаемости, что приведет к большему накоплению мутаций. Дрожжевые Rev1 и человеческая полимераза η являются членами транслезионных ДНК- полимераз семейства Y , присутствующих во время глобального ответа на повреждение ДНК и ответственных за усиленный мутагенез во время глобального ответа на повреждение ДНК у эукариот. [ 42 ]
Старение
[ редактировать ]Патологические последствия плохой репарации ДНК
[ редактировать ]
Экспериментальные животные с генетическими нарушениями репарации ДНК часто демонстрируют снижение продолжительности жизни и увеличение заболеваемости раком. [ 13 ] Например, мыши с дефицитом доминантного пути NHEJ и механизмов поддержания теломер чаще заболевают лимфомой и инфекциями и, как следствие, имеют более короткую продолжительность жизни, чем мыши дикого типа. [ 62 ] Аналогичным образом, у мышей с дефицитом ключевого белка репарации и транскрипции, который раскручивает спирали ДНК, преждевременно возникают заболевания, связанные со старением, и, как следствие, сокращается продолжительность жизни. [ 63 ] Однако не каждый дефицит репарации ДНК приводит к именно предсказанным эффектам; у мышей с дефицитом пути NER наблюдалось сокращение продолжительности жизни без соответствующего более высокого уровня мутаций. [ 64 ]
Максимальная продолжительность жизни мышей составляет соответственно ~3, ~ , голых землекопов и человека 30 и ~129 лет. [ 65 ] Из них самый короткоживущий вид, мышь, экспрессирует гены репарации ДНК, включая основные гены в нескольких путях репарации ДНК, на более низком уровне, чем у людей и голых землекопов. [ 65 ] Кроме того, некоторые пути репарации ДНК у людей и голых землекопов активируются по сравнению с мышами. Эти наблюдения позволяют предположить, что повышенная репарация ДНК способствует увеличению продолжительности жизни . [ 65 ]
Если скорость повреждения ДНК превышает способность клетки восстанавливать ее, накопление ошибок может сокрушить клетку и привести к раннему старению, апоптозу или раку. Наследственные заболевания, связанные с нарушением функции восстановления ДНК, приводят к преждевременному старению. [ 13 ] повышенная чувствительность к канцерогенам и, соответственно, повышенный риск развития рака (см. ниже ). С другой стороны, организмы с усиленными системами репарации ДНК, такие как Deinococcus radiodurans , вызывающим двухцепочечный разрыв , наиболее устойчивый к радиации известный организм, проявляют замечательную устойчивость к эффектам радиоактивности , вероятно, из-за повышенной эффективности репарации ДНК и особенно НХЭЖ. [ 66 ]
Продолжительность жизни и ограничение калорий
[ редактировать ]
Было обнаружено, что ряд отдельных генов влияют на вариации продолжительности жизни внутри популяции организмов. Эффекты этих генов сильно зависят от окружающей среды, в частности, от питания организма. Ограничение калорий воспроизводимо приводит к увеличению продолжительности жизни у различных организмов, вероятно, за счет путей восприятия питательных веществ и снижения скорости метаболизма . Молекулярные механизмы, с помощью которых такое ограничение приводит к увеличению продолжительности жизни, пока неясны (см. [ 67 ] для некоторого обсуждения); однако поведение многих генов, которые, как известно, участвуют в репарации ДНК, изменяется в условиях ограничения калорий. Было показано, что несколько агентов, обладающих антивозрастными свойствами, снижают конститутивный уровень передачи сигналов mTOR , что свидетельствует о снижении метаболической активности , и одновременно снижают конститутивный уровень повреждения ДНК , вызванного эндогенно генерируемыми активными формами кислорода. [ 68 ]
Например, увеличение дозировки гена SIR-2, который регулирует упаковку ДНК у нематодного червя Caenorhabditis elegans , может значительно продлить продолжительность жизни. [ 69 ] Известно, что гомолог SIR-2 млекопитающих индуцирует нижестоящие факторы репарации ДНК, участвующие в NHEJ, активность, которая особенно усиливается в условиях ограничения калорий. [ 70 ] Ограничение калорий тесно связано со скоростью репарации вырезаемых оснований в ядерной ДНК грызунов. [ 71 ] хотя подобные эффекты не наблюдались в митохондриальной ДНК. [ 72 ]
Ген AGE-1 C. elegans , вышестоящий эффектор путей восстановления ДНК, обеспечивает резкое увеличение продолжительности жизни в условиях свободного кормления, но приводит к снижению репродуктивной способности в условиях ограничения калорий. [ 73 ] Это наблюдение подтверждает плейотропную теорию биологических причин старения , которая предполагает, что гены, дающие большое преимущество в выживании в раннем возрасте, будут отбираться, даже если они несут соответствующий недостаток в более позднем возрасте.
Медицина и модуляция восстановления ДНК
[ редактировать ]Наследственные нарушения репарации ДНК
[ редактировать ]Дефекты в механизме NER ответственны за несколько генетических нарушений, в том числе:
- Пигментная ксеродерма : повышенная чувствительность к солнечному свету/УФ-излучению, что приводит к увеличению заболеваемости раком кожи и преждевременному старению.
- Синдром Кокейна : повышенная чувствительность к ультрафиолету и химическим агентам.
- Трихотиодистрофия : чувствительная кожа, ломкость волос и ногтей.
Умственная отсталость часто сопровождает два последних расстройства, что указывает на повышенную уязвимость нейронов развития.
Другие нарушения репарации ДНК включают:
- Синдром Вернера : преждевременное старение и задержка роста.
- Синдром Блума : гиперчувствительность к солнечному свету, высокая частота злокачественных новообразований (особенно лейкозов ).
- Атаксия телеангиэктазия : чувствительность к ионизирующему излучению и некоторым химическим агентам.
Все вышеперечисленные заболевания часто называют «сегментарными прогериями » (« болезнями ускоренного старения »), поскольку заболевшие выглядят пожилыми и испытывают возрастные заболевания в аномально молодом возрасте, при этом не проявляя всех симптомов старости.
Другие заболевания, связанные со снижением функции восстановления ДНК, включают анемию Фанкони , наследственный рак молочной железы и наследственный рак толстой кишки .
Рак
[ редактировать ]Из-за присущих механизмам восстановления ДНК ограничений, если бы люди жили достаточно долго, у всех них в конечном итоге развился бы рак. [ 74 ] [ 75 ] Существует по меньшей мере 34 наследственных мутации генов репарации ДНК человека, которые повышают риск развития рака . Многие из этих мутаций приводят к тому, что восстановление ДНК становится менее эффективным, чем обычно. В частности, наследственный неполипозный колоректальный рак (HNPCC) тесно связан со специфическими мутациями в пути восстановления несоответствия ДНК. BRCA1 и BRCA2 , два важных гена, мутации которых приводят к значительному увеличению риска рака молочной железы у носителей. [ 76 ] оба связаны с большим количеством путей репарации ДНК, особенно NHEJ и гомологичной рекомбинации.
Процедуры лечения рака, такие как химиотерапия и лучевая терапия , подавляют способность клеток восстанавливать повреждения ДНК, что приводит к их гибели. Предпочтительно поражаются клетки, которые делятся наиболее быстро – чаще всего раковые клетки. Побочным эффектом является то, что поражаются и другие нераковые, но быстро делящиеся клетки, такие как клетки-предшественники в кишечнике, коже и кроветворной системе. Современные методы лечения рака пытаются локализовать повреждение ДНК в клетках и тканях, связанных только с раком, либо физическими средствами (концентрация терапевтического агента в области опухоли), либо биохимическими средствами (используя особенность, уникальную для раковых клеток в организме). . В контексте терапии, нацеленной на гены, отвечающие за повреждение ДНК, последний подход получил название «синтетическая летальность». [ 77 ]
Пожалуй, самым известным из этих «синтетических летальных» препаратов является ингибитор поли(АДФ-рибозы) полимеразы 1 ( PARP1 ) олапариб , который был одобрен Управлением по контролю за продуктами и лекарствами в 2015 году для лечения женщин с дефектом яичников по BRCA. рак. Опухолевые клетки с частичной потерей реакции на повреждение ДНК (в частности, репарации гомологичной рекомбинации ) зависят от другого механизма – репарации одноцепочечных разрывов, который частично основан на продукте гена PARP1. [ 78 ] Олапариб комбинируется с химиотерапевтическими средствами для ингибирования восстановления одноцепочечных разрывов, вызванных повреждением ДНК, вызванным одновременным применением химиотерапии. Опухолевые клетки, опирающиеся на этот механизм репарации остаточной ДНК, не способны восстанавливать повреждения и, следовательно, не способны выживать и пролиферировать, тогда как нормальные клетки могут восстанавливать повреждения с помощью функционирующего механизма гомологичной рекомбинации.
В настоящее время исследуются многие другие препараты для использования против других остаточных механизмов репарации ДНК, обычно встречающихся при раке. Однако синтетические подходы к лечению летальности были поставлены под сомнение из-за появляющихся доказательств приобретенной устойчивости, достигаемой за счет перестройки путей ответа на повреждение ДНК и реверсии ранее подавленных дефектов. [ 79 ]
Дефекты репарации ДНК при раке
[ редактировать ]За последние несколько лет стало очевидно, что реакция на повреждение ДНК действует как барьер на пути злокачественной трансформации предопухолевых клеток. [ 80 ] Предыдущие исследования показали повышенную реакцию на повреждение ДНК в моделях клеточных культур с активацией онкогена. [ 81 ] и предопухолевые аденомы толстой кишки. [ 82 ] Механизмы ответа на повреждение ДНК запускают остановку клеточного цикла и пытаются восстановить повреждения ДНК или способствуют гибели/старению клеток, если восстановление невозможно. Стресс репликации наблюдается в предопухолевых клетках из-за усиления сигналов пролиферации от онкогенных мутаций. Репликативный стресс характеризуется: повышенным запуском инициации/оригинации репликации; усиление транскрипции и столкновение комплексов транскрипции-репликации; дефицит нуклеотидов; увеличение активных форм кислорода (АФК). [ 83 ]
Репликативный стресс, наряду с отбором инактивирующих мутаций в генах реакции на повреждение ДНК в эволюции опухоли, [ 84 ] приводит к подавлению и/или потере некоторых механизмов ответа на повреждение ДНК и, следовательно, к потере репарации ДНК и/или старению/запрограммированной гибели клеток. В экспериментальных моделях на мышах потеря клеточного старения, опосредованного реакцией на повреждение ДНК, наблюдалась после использования короткой шпилечной РНК (shRNA) для ингибирования киназы атаксии-телеангиэктазии ( АТМ ), что приводило к увеличению размера опухоли и ее инвазивности. [ 82 ] Люди, рожденные с наследственными дефектами механизмов репарации ДНК (например, синдром Ли-Фраумени ), имеют более высокий риск развития рака. [ 85 ]
Распространенность мутаций в ответ на повреждение ДНК различается в зависимости от типа рака; например, 30% инвазивных карцином молочной железы имеют мутации в генах, участвующих в гомологичной рекомбинации. [ 80 ] При раке снижение регуляции наблюдается во всех механизмах реакции на повреждение ДНК (восстановление вырезания оснований (BER), восстановление вырезания нуклеотидов (NER), восстановление несоответствия ДНК (MMR), репарация гомологичной рекомбинации (HR), негомологическое соединение концов (NHEJ) и транслезный синтез ДНК (TLS). [ 86 ] Помимо мутаций в генах восстановления повреждений ДНК, мутации также возникают в генах, ответственных за остановку клеточного цикла , чтобы дать достаточно времени для восстановления ДНК, а некоторые гены участвуют как в восстановлении повреждений ДНК, так и в контроле контрольных точек клеточного цикла, например ATM и киназа контрольной точки 2 (CHEK2) – супрессор опухоли, который часто отсутствует или снижается при немелкоклеточном раке легкого. [ 87 ]
| HR | НХЭЖ | ССА | НО | БЕР | ВНИЗ | ММР | |
|---|---|---|---|---|---|---|---|
| банкомат | |||||||
| ATR | |||||||
| ПАКСИП | |||||||
| РПА | |||||||
| БРЦА1 | |||||||
| БРЦА2 | |||||||
| РАД51 | |||||||
| RFC | |||||||
| XRCC1 | |||||||
| ПКНА | |||||||
| ПАРП1 | |||||||
| ERCC1 | |||||||
| МШ3 |
Эпигенетические дефекты репарации ДНК при раке
[ редактировать ]Классически рак рассматривается как совокупность заболеваний, вызванных прогрессирующими генетическими аномалиями, включающими мутации в генах-супрессорах опухолей и онкогенах, а также хромосомные аберрации. Однако стало очевидно, что рак также вызван эпигенетическими изменениями . [ 88 ]
Эпигенетические изменения относятся к функционально значимым модификациям генома, которые не связаны с изменением нуклеотидной последовательности. Примерами таких модификаций являются изменения метилирования ДНК (гиперметилирование и гипометилирование) и модификация гистонов . [ 89 ] изменения в хромосомной архитектуре (вызванные несоответствующей экспрессией белков, таких как HMGA2 или HMGA1 ) [ 90 ] и изменения, вызванные микроРНК . Каждое из этих эпигенетических изменений служит для регуляции экспрессии генов без изменения базовой последовательности ДНК . Эти изменения обычно сохраняются в результате деления клеток , длятся несколько поколений клеток и могут рассматриваться как эпимутации (эквивалентные мутациям).
Хотя при раке обнаруживается большое количество эпигенетических изменений, эпигенетические изменения в генах репарации ДНК, вызывающие снижение экспрессии белков репарации ДНК, кажутся особенно важными. Считается, что такие изменения происходят на ранних стадиях развития рака и являются вероятной причиной генетической нестабильности, характерной для рака. [ 91 ] [ 92 ] [ 93 ]
Снижение экспрессии генов репарации ДНК приводит к недостаточной репарации ДНК. Когда репарация ДНК недостаточна, повреждения ДНК остаются в клетках на более высоком, чем обычно, уровне, и эти избыточные повреждения вызывают повышенную частоту мутаций или эпимутаций. Частота мутаций существенно увеличивается в клетках, дефектных в репарации несоответствий ДНК. [ 94 ] [ 95 ] или в гомологичной рекомбинационной репарации (HRR). [ 96 ] Хромосомные перестройки и анеуплоидия также увеличиваются в клетках с дефектами HRR. [ 97 ]
Более высокие уровни повреждения ДНК не только вызывают усиление мутаций, но и усиление эпимутации. Во время репарации двухцепочечных разрывов ДНК или других повреждений ДНК не полностью очищенные участки репарации могут вызывать эпигенетическое молчание генов. [ 98 ] [ 99 ]
Недостаточная экспрессия белков репарации ДНК из-за наследственной мутации может вызвать повышенный риск развития рака. Лица с унаследованным нарушением любого из 34 генов репарации ДНК (см. статью « Расстройство, связанное с дефицитом репарации ДНК ») имеют повышенный риск развития рака, при этом некоторые дефекты вызывают до 100% вероятность возникновения рака на протяжении всей жизни (например, мутации p53). [ 100 ] Однако такие мутации зародышевой линии (которые вызывают синдромы высокопроникающего рака) являются причиной лишь около 1 процента случаев рака. [ 101 ]
Частоты эпимутаций в генах репарации ДНК
[ редактировать ]
Дефицит ферментов репарации ДНК иногда вызван вновь возникающей соматической мутацией в гене репарации ДНК, но гораздо чаще вызван эпигенетическими изменениями, которые уменьшают или подавляют экспрессию генов репарации ДНК. Например, когда последовательно исследовали 113 случаев колоректального рака, только четыре имели миссенс-мутацию в гене репарации ДНК MGMT , в то время как у большинства наблюдалось снижение экспрессии MGMT из-за метилирования области промотора MGMT (эпигенетическое изменение). [ 102 ] Пять различных исследований показали, что от 40% до 90% случаев колоректального рака снижают экспрессию MGMT из-за метилирования области промотора MGMT. [ 103 ] [ 104 ] [ 105 ] [ 106 ] [ 107 ]
Аналогичным образом, из 119 случаев колоректального рака с дефицитом репарации ошибочного спаривания, при котором отсутствовала экспрессия гена репарации ДНК PMS2 , PMS2 был дефицитным в 6 из-за мутаций в гене PMS2, тогда как в 103 случаях экспрессия PMS2 была недостаточной, потому что его партнер по спариванию MLH1 был подавлен из-за метилированию промотора (белок PMS2 нестабильен в отсутствие MLH1). [ 108 ] В других 10 случаях потеря экспрессии PMS2, вероятно, была связана с эпигенетической сверхэкспрессией микроРНК , miR -155 , которая подавляет MLH1. [ 109 ]
В следующем примере эпигенетические дефекты были обнаружены при различных видах рака (например, рака молочной железы, яичников, колоректального рака, а также рака головы и шеи). Два или три дефицита экспрессии ERCC1 , XPF или PMS2 встречаются одновременно в большинстве из 49 случаев рака толстой кишки, оцененных Facista et al. [ 110 ]
В таблице в этом разделе показаны некоторые часто встречающиеся агенты, повреждающие ДНК, примеры повреждений ДНК, которые они вызывают, и пути, которые устраняют эти повреждения ДНК. По крайней мере 169 ферментов либо непосредственно участвуют в репарации ДНК, либо влияют на процессы репарации ДНК. [ 111 ] Из них 83 непосредственно задействованы в восстановлении 5 типов повреждений ДНК, показанных на диаграмме. [ нужна ссылка ]
Некоторые из наиболее хорошо изученных генов, играющих центральную роль в процессах восстановления, показаны на диаграмме. Обозначения генов, выделенные красным, серым или голубым цветом, указывают на гены, которые часто эпигенетически изменяются при различных типах рака. Статьи в Википедии о каждом гене, выделенном красным, серым или голубым, описывают эпигенетические изменения и рак, при которых обнаруживаются эти эпимутации. Обзорные статьи, [ 112 ] и обширные экспериментальные обзорные статьи [ 113 ] [ 114 ] также документируют большинство этих нарушений репарации эпигенетической ДНК при раке.
Гены, выделенные красным, часто редуцируются или подавляются эпигенетическими механизмами при различных видах рака. Когда экспрессия этих генов низкая или отсутствует, повреждения ДНК могут накапливаться. Ошибки репликации, выходящие за пределы этих повреждений (см. Синтез транслезий ), могут привести к увеличению количества мутаций и, в конечном итоге, к раку. Эпигенетическая репрессия генов репарации ДНК в точных путях репарации ДНК, по-видимому, играет центральную роль в канцерогенезе .
Два выделенных серым гена RAD51 и BRCA2 необходимы для гомологичной рекомбинационной репарации. Иногда они эпигенетически сверхэкспрессируются, а иногда и недостаточно экспрессируются при некоторых видах рака. Как указано в статьях Википедии о RAD51 и BRCA2 , такие виды рака обычно имеют эпигенетический дефицит других генов репарации ДНК. Эти недостатки репарации, вероятно, приведут к увеличению количества невосстановленных повреждений ДНК. Сверхэкспрессия RAD51 и BRCA2, наблюдаемая при этих видах рака, может отражать селективное давление на компенсаторную RAD51 или BRCA2 сверхэкспрессию и усиление гомологичной рекомбинационной репарации, чтобы, по крайней мере, частично справиться с такими избыточными повреждениями ДНК. В тех случаях, когда RAD51 или BRCA2 недостаточно экспрессируются, это само по себе может привести к увеличению невосстановленных повреждений ДНК. Ошибки репликации, выходящие за рамки этих повреждений (см. «Синтез транслезий »), могут вызвать усиление мутаций и рак, так что недостаточная экспрессия RAD51 или BRCA2 сама по себе будет канцерогенной.
Гены, выделенные голубым цветом, участвуют в пути соединения концов, опосредованном микрогомологией (MMEJ), и их активация повышается при раке. MMEJ — это дополнительный, подверженный ошибкам и неточный путь восстановления двухцепочечных разрывов. При репарации двухцепочечного разрыва MMEJ гомология 5–25 комплементарных пар оснований между обеими парными цепями достаточна для выравнивания цепей, но обычно присутствуют несовпадающие концы (лоскуты). MMEJ удаляет лишние нуклеотиды (лоскуты) в местах соединения цепей, а затем лигирует нити, чтобы создать неповрежденную двойную спираль ДНК. MMEJ почти всегда включает хотя бы небольшую делецию, так что это мутагенный путь. [ 24 ] FEN1 , эндонуклеаза лоскута в MMEJ, эпигенетически увеличивается за счет гипометилирования промотора и сверхэкспрессируется в большинстве случаев рака молочной железы. [ 115 ] простата, [ 116 ] желудок, [ 117 ] [ 118 ] нейробластомы, [ 119 ] поджелудочная железа, [ 120 ] и легкое. [ 121 ] PARP1 также сверхэкспрессируется, когда ETS сайт его промоторной области эпигенетически гипометилирован, и это способствует прогрессированию рака эндометрия. [ 122 ] и серозный рак яичников с мутацией BRCA. [ 123 ] Другие гены пути MMEJ также сверхэкспрессируются при ряде видов рака (краткую информацию см. в MMEJ ) и также показаны голубым цветом.
Полногеномное распределение репарации ДНК в соматических клетках человека
[ редактировать ]Дифференциальная активность путей репарации ДНК в различных областях генома человека приводит к тому, что мутации распределяются внутри опухолевых геномов очень неравномерно. [ 124 ] [ 125 ] В частности, богатые генами и рано реплицирующиеся области человеческого генома демонстрируют более низкую частоту мутаций, чем бедный генами и поздно реплицирующийся гетерохроматин . Один из механизмов, лежащих в основе этого, включает модификацию гистонов H3K36me3 , которая может рекрутировать репарации несоответствий . белки [ 126 ] тем самым снижая частоту мутаций в областях, отмеченных H3K36me3 . [ 127 ] Другой важный механизм касается эксцизионной репарации нуклеотидов , которая может быть задействована транскрипционным аппаратом, снижая частоту соматических мутаций в активных генах. [ 125 ] и другие открытые области хроматина. [ 128 ]
Эпигенетические изменения вследствие репарации ДНК
[ редактировать ]Повреждения ДНК очень распространены и постоянно восстанавливаются. Эпигенетические изменения могут сопровождать восстановление ДНК в результате окислительного повреждения или двухцепочечных разрывов. В клетках человека окислительное повреждение ДНК происходит примерно 10 000 раз в день, а двухцепочечные разрывы ДНК происходят примерно от 10 до 50 раз за клеточный цикл в соматических реплицирующихся клетках (см. Повреждение ДНК (естественное происхождение) ). Избирательное преимущество репарации ДНК заключается в том, что она позволяет клетке выжить даже после повреждения ДНК. Избирательное преимущество эпигенетических изменений, возникающих при репарации ДНК, неясно. [ нужна ссылка ]
Восстановление окислительных повреждений ДНК может изменить эпигенетические маркеры
[ редактировать ]В устойчивом состоянии (с возникновением и восстановлением эндогенных повреждений) имеется около 2400 окислительно поврежденных гуанинов, которые образуют 8-оксо-2'-дезоксигуанозин (8-OHdG). в средней ДНК клеток млекопитающих [ 129 ] 8-OHdG составляет около 5% окислительных повреждений, обычно присутствующих в ДНК. [ 130 ] Окисленные гуанины не встречаются случайно среди всех гуанинов в ДНК. Существует предпочтение последовательности гуанина в метилированном сайте CpG (цитозин, за которым следует гуанин в направлении 5' → 3' и где цитозин метилирован (5-mCpG)). [ 131 ] Сайт 5-mCpG имеет самый низкий потенциал ионизации для окисления гуанина.

Окисленный гуанин обладает потенциалом неправильного спаривания и является мутагенным. [ 133 ] Оксогуанингликозилаза (OGG1) является основным ферментом, ответственным за удаление окисленного гуанина во время репарации ДНК. OGG1 находит 8-OHdG и связывается с ним в течение нескольких секунд. [ 134 ] Однако OGG1 не удаляет 8-OHdG немедленно. В клетках HeLa половина максимального удаления 8-OHdG происходит за 30 минут. [ 135 ] а у облученных мышей 8-OHdG, индуцированные в печени мышей, удаляются с периодом полураспада 11 минут. [ 130 ]
Когда OGG1 присутствует в окисленном гуанине в метилированном сайте CpG, он привлекает TET1 к поражению 8-OHdG (см. Рисунок). Это позволяет TET1 деметилировать соседний метилированный цитозин. [ 136 ] Деметилирование цитозина является эпигенетическим изменением. [ 137 ]
Например, когда эпителиальные клетки молочной железы человека обрабатывались H 2 O 2 в течение шести часов, содержание 8-OHdG в ДНК увеличивалось примерно в 3,5 раза, что вызывало примерно 80% деметилирование 5-метилцитозинов в геноме. [ 132 ] Деметилирование CpG в промоторе гена под действием фермента TET увеличивает транскрипцию гена в информационную РНК. [ 138 ] В клетках, обработанных H 2 O 2 , исследовали один конкретный ген, BACE1 . [ 132 ] Уровень метилирования BACE1 CpG-островка был снижен (эпигенетическое изменение), что позволило примерно в 6,5 раз увеличить экспрессию информационной РНК BACE1 . [ нужна ссылка ]
В то время как шестичасовая инкубация с H 2 O 2 вызывает значительное деметилирование сайтов 5-mCpG, более короткое время инкубации с H 2 O 2 , по-видимому, способствует другим эпигенетическим изменениям. Обработка клеток H 2 O 2 в течение 30 минут приводит к тому, что гетеродимер белка репарации несоответствия MSH2-MSH6 рекрутирует ДНК-метилтрансферазу 1 (DNMT1) в места некоторых видов окислительного повреждения ДНК. [ 139 ] Это может вызвать повышенное метилирование цитозинов (эпигенетические изменения) в этих местах.
Цзян и др. [ 140 ] обрабатывали клетки HEK 293 агентами, вызывающими окислительное повреждение ДНК, ( броматом калия (KBrO3) или хроматом калия (K2CrO4)). Базовая эксцизионная репарация (BER) окислительного повреждения происходила с помощью фермента репарации ДНК- полимеразы бета, локализующегося на окисленных гуанинах. Полимераза бета является основной полимеразой человека при коротком BER окислительного повреждения ДНК. Цзян и др. [ 140 ] также обнаружили, что полимераза бета рекрутирует белок ДНК-метилтрансферазы DNMT3b в сайты репарации BER. Затем они оценили характер метилирования на уровне одного нуклеотида в небольшой области ДНК, включая область промотора и область ранней транскрипции гена BRCA1 . Окислительное повреждение ДНК броматом модулировало характер метилирования ДНК (вызывая эпигенетические изменения) в сайтах CpG в исследуемой области ДНК. В необработанных клетках CpG, расположенные в местах -189, -134, -29, -19, +16 и +19 гена BRCA1, содержали метилированные цитозины (где нумерация ведется от места начала транскрипции информационной РНК , а отрицательные числа указывают на нуклеотиды в вышележащая промоторная область). Окисление, вызванное обработкой броматом, привело к потере метилирования цитозина в точках -189, -134, +16 и +19, а также привело к образованию нового метилирования в CpG, расположенных в точках -80, -55, -21 и +8 после Репарация ДНК была разрешена. [ нужна ссылка ]
Гомологичная рекомбинационная репарация изменяет эпигенетические маркеры
[ редактировать ]По крайней мере в четырех статьях сообщается о привлечении ДНК-метилтрансферазы 1 (DNMT1) к участкам двухцепочечных разрывов ДНК. [ 141 ] [ 142 ] [ 98 ] [ 143 ] Во время гомологичной рекомбинационной репарации (HR) двухцепочечного разрыва участие DNMT1 приводит к тому, что две восстановленные цепи ДНК имеют разные уровни метилированных цитозинов. Одна цепь часто метилируется примерно в 21 сайте CpG ниже восстановленного двухцепочечного разрыва. Другая цепь ДНК теряет метилирование примерно в шести сайтах CpG, которые ранее были метилированы после двухцепочечного разрыва, а также теряет метилирование примерно в пяти сайтах CpG, которые ранее были метилированы перед двухцепочечным разрывом. Когда хромосома реплицируется, это приводит к появлению одной дочерней хромосомы, которая сильно метилирована ниже предыдущего сайта разрыва, и одной, которая неметилирована в области как выше, так и ниже предыдущего сайта разрыва. Что касается гена, который был сломан в результате двухцепочечного разрыва, половина клеток-потомков экспрессирует этот ген на высоком уровне, а в другой половине клеток-потомков экспрессия этого гена подавляется. Когда клоны этих клеток хранились в течение трех лет, новые закономерности метилирования сохранялись в течение этого периода времени. [ 144 ]
У мышей с CRISPR-опосредованной вставкой рекомбинации, направленной на гомологию, в их геноме наблюдалось большое количество повышенного метилирования сайтов CpG внутри вставки, связанной с двухцепочечным разрывом. [ 145 ]
Негомологичное соединение концов может вызывать некоторые изменения эпигенетических маркеров.
[ редактировать ]негомологичным соединением концов (NHEJ) может вызвать небольшое количество деметилирований ранее существовавших метилирований цитозиновой ДНК ниже восстановленного двухцепочечного разрыва. Восстановление двухцепочечного разрыва [ 142 ] Дальнейшая работа Аллена и др. [ 146 ] показали, что NHEJ двухцепочечного разрыва ДНК в клетке может давать начало некоторым клеткам-потомкам, имеющим репрессированную экспрессию гена, несущего первоначальный двухцепочечный разрыв, и некоторому потомству, имеющему высокую экспрессию этого гена из-за эпигенетических изменений, связанных с репарацией NHEJ. . Частота эпигенетических изменений, вызывающих репрессию гена после репарации NHEJ двухцепочечного разрыва ДНК в этом гене, может составлять около 0,9%. [ 98 ]
Эволюция
[ редактировать ]Основные процессы репарации ДНК высоко консервативны как у прокариот , так и у эукариот и даже у бактериофагов ( вирусов , поражающих бактерии ); однако более сложные организмы с более сложными геномами имеют соответственно более сложные механизмы восстановления. [ 147 ] Способность большого числа структурных мотивов белков катализировать соответствующие химические реакции сыграла значительную роль в разработке механизмов репарации в ходе эволюции. Чрезвычайно подробный обзор гипотез, касающихся эволюции репарации ДНК, см. [ 148 ]
Летопись окаменелостей указывает на то, что одноклеточная жизнь начала распространяться на планете в какой-то момент в докембрийский период, хотя точно неясно, когда именно возникла современная жизнь. Нуклеиновые кислоты стали единственным и универсальным средством кодирования генетической информации, требующим механизмов репарации ДНК, которые в своей основной форме были унаследованы всеми существующими формами жизни от их общего предка. Появление на Земле богатой кислородом атмосферы (известной как « кислородная катастрофа ») из-за фотосинтезирующих организмов, а также наличие потенциально повреждающих свободных радикалов в клетке из-за окислительного фосфорилирования , вызвало необходимость эволюции механизмов репарации ДНК, действующих специфическим образом. для противодействия типам повреждений, вызванных окислительным стрессом . Однако механизм, благодаря которому это произошло, неясен. [ нужна ссылка ]
Скорость эволюционных изменений
[ редактировать ]В некоторых случаях повреждение ДНК не восстанавливается или восстанавливается с помощью механизма, подверженного ошибкам, что приводит к изменению исходной последовательности. Когда это происходит, мутации могут распространиться на геномы потомства клетки. Если такое событие произойдет в клетке зародышевой линии , которая в конечном итоге продуцирует гамету , мутация может быть передана потомству организма. Скорость эволюции конкретного вида (или конкретного гена) является функцией скорости мутаций. Как следствие, скорость и точность механизмов репарации ДНК оказывают влияние на процесс эволюционных изменений. [ 149 ] Защита и восстановление повреждений ДНК не влияют на скорость адаптации за счет регуляции генов, а также за счет рекомбинации и отбора аллелей. С другой стороны, восстановление и защита повреждений ДНК действительно влияет на скорость накопления непоправимых, полезных, расширяющих код наследственных мутаций и замедляет эволюционный механизм расширения генома организмов с новыми функциональными возможностями. Противоречие между эволюционируемостью и восстановлением и защитой мутаций требует дальнейшего изучения. [ нужна ссылка ]
Технология
[ редактировать ]В 2012 году была открыта технология, получившая название «Кластеризованные короткие палиндромные повторы с регулярными промежутками» (сокращенно CRISPR -Cas9). Новая технология позволяет любому человеку, имеющему образование в области молекулярной биологии, с точностью изменять гены любого вида, вызывая повреждение ДНК в определенной точке, а затем изменение механизмов репарации ДНК для вставки новых генов. [ 150 ] Это дешевле, эффективнее и точнее, чем другие технологии. С помощью CRISPR-Cas9 ученые могут редактировать части генома, удаляя, добавляя или изменяя части последовательности ДНК. [ нужна ссылка ]
См. также
[ редактировать ]- Болезнь ускоренного старения
- Старение ДНК
- Клеточный цикл
- Повреждение ДНК (естественное)
- Теория старения, связанная с повреждением ДНК
- репликация ДНК
- Прямое повреждение ДНК
- Обнаружение и исправление ошибок
- Генная терапия
- Митохондриальная генетика человека
- Косвенное повреждение ДНК
- Продление жизни
- Прогерия
- РЕМОНТ
- Старение
- СиДНК
- Научный журнал « Репарация ДНК в условиях исследования мутаций»
Ссылки
[ редактировать ]- ^ «Серия обзоров природы: повреждение ДНК» . Nature Reviews Молекулярно-клеточная биология . 5 июля 2017 года . Проверено 7 ноября 2018 г.
- ^ Jump up to: а б Лодиш Х., Берк А., Мацудайра П., Кайзер К.А., Кригер М., Скотт М.П. и др. (2004). Молекулярно-клеточная биология (5-е изд.). У. Х. Фриман. п. 963. ИСБН 978-0-7167-4366-8 . OCLC 53798180 .
- ^ Браунер В.С., Кан А.Дж., Зив Э., Райнер А.П., Осима Дж., Коутон Р.М. и др. (декабрь 2004 г.). «Генетика долголетия человека». Американский медицинский журнал . 117 (11): 851–60. CiteSeerX 10.1.1.556.6874 . doi : 10.1016/j.amjmed.2004.06.033 . ПМИД 15589490 .
- ^ Broad WJ (7 октября 2015 г.). «Нобелевская премия по химии присуждена Томасу Линдалю, Полу Модричу и Азизу Санкару за исследования ДНК» . Нью-Йорк Таймс . Проверено 7 октября 2015 г.
- ^ Персонал (7 октября 2015 г.). «Нобелевская премия по химии 2015 г. – восстановление ДНК – обеспечение химической стабильности на всю жизнь» (PDF) . Нобелевская премия . Проверено 7 октября 2015 г.
- ^ Роулстон А., Марселлус Р.К., Брэнтон П.Е. (1999). «Вирусы и апоптоз». Ежегодный обзор микробиологии . 53 : 577–628. дои : 10.1146/аннурев.микро.53.1.577 . ПМИД 10547702 .
- ^ Мэдиган М.Т., Мартино Дж.М. (2006). Брок Биология микроорганизмов (11-е изд.). Пирсон. п. 136. ИСБН 978-0-13-196893-6 .
- ^ Охта Т., Токишита С.И., Мотидзуки К., Кавасе Дж., Сакахира М., Ямагата Х. (2006). «Чувствительность к УФ-излучению и мутагенез чрезвычайно термофильной Eubacterium Thermus thermophilus HB27» . Гены и окружающая среда . 28 (2): 56–61. Бибкод : 2006GeneE..28...56O . дои : 10.3123/jemsge.28.56 .
- ^ Танака Т., Халица Х.Д., Хуанг Х., Траганос Ф., Даржинкевич З. (сентябрь 2006 г.). «Конститутивное фосфорилирование гистона H2AX и активация АТМ, репортеры повреждения ДНК эндогенными оксидантами» . Клеточный цикл . 5 (17): 1940–45. дои : 10.4161/cc.5.17.3191 . ПМК 3488278 . ПМИД 16940754 .
- ^ Брэйг М., Шмитт, Калифорния (март 2006 г.). «Онкоген-индуцированное старение: тормозить развитие опухоли» . Исследования рака . 66 (6): 2881–4. дои : 10.1158/0008-5472.CAN-05-4006 . ПМИД 16540631 .
- ^ Линч, доктор медицинских наук (февраль 2006 г.). «Как клеточное старение предотвращает рак?». ДНК и клеточная биология . 25 (2): 69–78. дои : 10.1089/dna.2006.25.69 . ПМИД 16460230 .
- ^ Кампизи Дж., Д'Адда ди Фаганья Ф (сентябрь 2007 г.). «Клеточное старение: когда с хорошими клетками случаются плохие вещи». Обзоры природы. Молекулярно-клеточная биология . 8 (9): 729–40. дои : 10.1038/nrm2233 . ПМИД 17667954 . S2CID 15664931 .
- ^ Jump up to: а б с Лучший БП (июнь 2009 г.). «Повреждение ядерной ДНК как прямая причина старения» (PDF) . Исследования омоложения . 12 (3): 199–208. CiteSeerX 10.1.1.318.738 . дои : 10.1089/rej.2009.0847 . ПМИД 19594328 . Архивировано из оригинала (PDF) 15 ноября 2017 года . Проверено 29 сентября 2009 г.
- ^ Санджар А (июнь 2003 г.). «Структура и функция ДНК-фотолиазы и криптохромных фоторецепторов синего света». Химические обзоры . 103 (6): 2203–37. дои : 10.1021/cr0204348 . ПМИД 12797829 .
- ^ Лукас-Льедо Дж.И., Линч М. (май 2009 г.). «Эволюция частоты мутаций: филогеномный анализ семейства фотолиаз/криптохромов» . Молекулярная биология и эволюция . 26 (5): 1143–53. дои : 10.1093/molbev/msp029 . ПМЦ 2668831 . ПМИД 19228922 .
- ^ Jump up to: а б с Уотсон Дж.Д., Бейкер Т.А., Белл С.П., Ганн А., Левин М., Лосик Р. (2004). Молекулярная биология гена (5-е изд.). Пирсон Бенджамин Каммингс; ЦШЛ Пресс. Ч. 9, 10. OCLC 936762772 .
- ^ Волкерт М.Р. (1988). «Адаптивный ответ Escherichia coli на повреждение алкилирования». Экологический и молекулярный мутагенез . 11 (2): 241–55. Бибкод : 1988EnvMM..11..241V . дои : 10.1002/em.2850110210 . ПМИД 3278898 . S2CID 24722637 .
- ^ Jump up to: а б с Уилли Дж., Шервуд Л., Вулвертон С. (2014). Микробиология Прескотта . Нью-Йорк: МакГроу Хилл. п. 381. ИСБН 978-0-07-340240-6 .
- ^ Рассел П. (2018). я генетика . Ченнаи: Пирсон. п. 186. ИСБН 978-93-325-7162-4 .
- ^ Jump up to: а б с д Рирдон Дж. Т., Санкар А. (2006). «Очистка и характеристика ферментных систем Escherichia coli и эксцизионной репарации нуклеотидов человека». Восстановление ДНК, Часть А. Методы энзимологии. Том. 408. стр. 189–213. дои : 10.1016/S0076-6879(06)08012-8 . ISBN 978-0-12-182813-4 . ПМИД 16793370 .
- ^ Берг М., Тимочко Дж., Страйер Л. (2012). Биохимия 7-е издание . Нью-Йорк: WH Freeman and Company. п. 840. ИСБН 978-1-4292-2936-4 .
- ^ Ачарья П.В. (1971). «Выделение и частичная характеристика коррелирующих с возрастом олигодезоксириборибонуклеотидов с ковалентно связанными аспартил-глутамил-полипептидами». Медицинский журнал Джонса Хопкинса. Приложение (1): 254–60. ПМИД 5055816 .
- ^ Бьоркстен Дж., Ачарья П.В., Ашман С., Ветлауфер Д.Б. (июль 1971 г.). «Герогенные фракции у тритированной крысы». Журнал Американского гериатрического общества . 19 (7): 561–74. дои : 10.1111/j.1532-5415.1971.tb02577.x . ПМИД 5106728 . S2CID 33154242 .
- ^ Jump up to: а б Лян Л., Дэн Л., Чен Ю., Ли Г.К., Шао С., Тишфилд Дж.А. (сентябрь 2005 г.). «Модуляция соединения концов ДНК ядерными белками» . Журнал биологической химии . 280 (36): 31442–49. дои : 10.1074/jbc.M503776200 . ПМИД 16012167 .
- ^ Уилсон Т.Э., Гравундер Ю., Либер М.Р. (июль 1997 г.). «Дренажная ДНК-лигаза IV опосредует негомологическое соединение концов ДНК» . Природа . 388 (6641): 495–8. Бибкод : 1997Natur.388..495W . дои : 10.1038/41365 . ПМИД 9242411 . S2CID 4422938 .
- ^ Мур Дж.К., Хабер Дж.Э. (май 1996 г.). «Клеточный цикл и генетические требования двух путей негомологичного восстановления двухцепочечных разрывов с соединением концов у Saccharomyces cerevisiae» . Молекулярная и клеточная биология . 16 (5): 2164–73. дои : 10.1128/mcb.16.5.2164 . ПМК 231204 . ПМИД 8628283 .
- ^ Бултон С.Дж., Джексон С.П. (сентябрь 1996 г.). «Saccharomyces cerevisiae Ku70 усиливает незаконную репарацию двухцепочечных разрывов ДНК и служит барьером для подверженных ошибкам путей репарации ДНК» . Журнал ЭМБО . 15 (18): 5093–103. дои : 10.1002/j.1460-2075.1996.tb00890.x . ПМЦ 452249 . ПМИД 8890183 .
- ^ Уилсон Т.Е., Либер М.Р. (август 1999 г.). «Эффективный процессинг концов ДНК во время соединения негомологичных концов дрожжей. Доказательства пути, зависимого от ДНК-полимеразы бета (Pol4)» . Журнал биологической химии . 274 (33): 23599–609. дои : 10.1074/jbc.274.33.23599 . ПМИД 10438542 .
- ^ Будман Дж., Чу Дж. (февраль 2005 г.). «Обработка ДНК для негомологичного соединения концов бесклеточным экстрактом» . Журнал ЭМБО . 24 (4): 849–60. дои : 10.1038/sj.emboj.7600563 . ПМК 549622 . ПМИД 15692565 .
- ^ Ван Х, Перро А.Р., Такеда Ю, Цинь В, Ван Х, Илиакис Г (сентябрь 2003 г.). «Биохимические доказательства Ku-независимых резервных путей NHEJ» . Исследования нуклеиновых кислот . 31 (18): 5377–88. дои : 10.1093/нар/gkg728 . ПМК 203313 . ПМИД 12954774 . (В настоящее время в этом документе выражается обеспокоенность , см. дои : 10.1093/nar/gkaa228 , ПМИД 32239214 )
- ^ Юнг Д., Alt FW (январь 2004 г.). «Раскрытие рекомбинации V(D)J; понимание регуляции генов» . Клетка . 116 (2): 299–311. дои : 10.1016/S0092-8674(04)00039-X . ПМИД 14744439 . S2CID 16890458 .
- ^ Jump up to: а б Труонг Л.Н., Ли Й., Ши Л.З., Хван П.Ю., Хе Дж., Ван Х. и др. (май 2013 г.). «Соединение концов, опосредованное микрогомологией, и гомологичная рекомбинация разделяют начальный этап резекции концов для восстановления двухцепочечных разрывов ДНК в клетках млекопитающих» . Труды Национальной академии наук Соединенных Штатов Америки . 110 (19): 7720–5. Бибкод : 2013PNAS..110.7720T . дои : 10.1073/pnas.1213431110 . ПМЦ 3651503 . ПМИД 23610439 .
- ^ Шарма С., Джавадекар С.М., Пандей М., Шривастава М., Кумари Р., Рагхаван С.К. (март 2015 г.). «Гомология и ферментативные требования к зависимому от микрогомологии альтернативному соединению концов» . Смерть клеток и болезни . 6 (3): e1697. дои : 10.1038/cddis.2015.58 . ПМЦ 4385936 . ПМИД 25789972 .
- ^ Декоттиньи А (2013). «Альтернативные механизмы соединения концов: историческая перспектива» . Границы генетики . 4 : 48. дои : 10.3389/fgene.2013.00048 . ПМЦ 3613618 . ПМИД 23565119 .
- ^ Заградка К., Слэйд Д., Бэйлоне А., Соммер С., Авербек Д., Петранович М. и др. (октябрь 2006 г.). «Повторная сборка разрушенных хромосом у Deinococcus radiodurans». Природа . 443 (7111): 569–73. Бибкод : 2006Natur.443..569Z . дои : 10.1038/nature05160 . ПМИД 17006450 . S2CID 4412830 .
- ^ Стенерлёв Б., Карлссон К.Х., Купер Б., Ридберг Б. «Измерение мгновенных двухцепочечных разрывов ДНК в клетках млекопитающих без включения термолабильных участков: результаты для клеток, лишенных негомологичного соединения концов». Радиат Рез. Апрель 2003 г.; 159 (4): 502–10. doi : 10.1667/0033-7587(2003)159[0502:mopdds 2.0.co;2] ПМИД 12643795 .
- ^ Уотерс Л.С., Майнсингер Б.К., Уилтраут М.Э., Д'Суза С., Вудрафф Р.В., Уокер Г.К. (март 2009 г.). «Эукариотические транслезионные полимеразы, их роль и регуляция в устойчивости к повреждениям ДНК» . Обзоры микробиологии и молекулярной биологии . 73 (1): 134–54. дои : 10.1128/MMBR.00034-08 . ПМЦ 2650891 . ПМИД 19258535 .
- ^ Колис Л.К., Райчаудхури П., Басу А.К. (август 2008 г.). «Мутационная специфичность внутрицепочечных перекрестных связей гуанин-тимин и тимин-гуанин, индуцированных гамма-излучением, в клетках млекопитающих и синтез транслейкоза после повреждения гуанин-тимина человеческой ДНК-полимеразой эта» . Биохимия . 47 (31): 8070–9. дои : 10.1021/bi800529f . ПМЦ 2646719 . ПМИД 18616294 .
- ^ Райчаудхури П., Басу А.К. (март 2011 г.). «Генетическая потребность в мутагенезе перекрестной связи G[8,5-Me]T в Escherichia coli: ДНК-полимеразы IV и V конкурируют за рискованный обходной путь» . Биохимия . 50 (12): 2330–8. дои : 10.1021/bi102064z . ПМК 3062377 . ПМИД 21302943 .
- ^ «Транслезионный синтез» . Research.chem.psu.edu. Архивировано из оригинала 10 марта 2012 года . Проверено 14 августа 2012 г.
- ^ Ван Цзы (июль 2001 г.). «Синтез транслезий с помощью ДНК-полимераз семейства UmuC». Мутационные исследования . 486 (2): 59–70. дои : 10.1016/S0921-8777(01)00089-1 . ПМИД 11425512 .
- ^ Jump up to: а б с Фридберг Э.К., Уокер Г.К., Сиде В., Вуд Р.Д., Шульц Р.А., Элленбергер Т. (2006). Репарация ДНК и мутагенез, часть 3. АСМ Пресс. 2-е изд.
- ^ Лю Б, Ип РК, Чжоу Цз (ноябрь 2012 г.). «Ремоделирование хроматина, восстановление повреждений ДНК и старение» . Современная геномика . 13 (7): 533–47. дои : 10.2174/138920212803251373 . ПМЦ 3468886 . ПМИД 23633913 .
- ^ Халицка Х.Д., Чжао Х., Подгорецка М., Траганос Ф., Даржинкевич З. (июль 2009 г.). «Цитометрическое обнаружение релаксации хроматина — ранний показатель реакции на повреждение ДНК» . Клеточный цикл . 8 (14): 2233–7. дои : 10.4161/cc.8.14.8984 . ПМЦ 3856216 . ПМИД 19502789 .
- ^ Jump up to: а б с Селлу Х., Лебопен Т., Шапюи С., Смит Р., Хегеле А., Сингх Х.Р. и др. (декабрь 2016 г.). «Поли(АДФ-рибоза)-зависимый ремодератор хроматина Alc1 вызывает локальную релаксацию хроматина при повреждении ДНК» . Молекулярная биология клетки . 27 (24): 3791–9. дои : 10.1091/mbc.E16-05-0269 . ПМК 5170603 . ПМИД 27733626 .
- ^ Jump up to: а б Ван Метер М., Саймон М., Томблайн Дж., Мэй А., Морелло Т.Д., Хаббард Б.П. и др. (сентябрь 2016 г.). «JNK фосфорилирует SIRT6, чтобы стимулировать восстановление двухцепочечных разрывов ДНК в ответ на окислительный стресс путем привлечения PARP1 к разрывам ДНК» . Отчеты по ячейкам . 16 (10): 2641–50. дои : 10.1016/j.celrep.2016.08.006 . ПМК 5089070 . ПМИД 27568560 .
- ^ Jump up to: а б Хейнс Дж.Ф., Макдональд Д., Родриг А., Дери Ю., Массон Дж.Ю., Хендзель М.Дж. и др. (январь 2008 г.). «PARP1-зависимая кинетика привлечения белков MRE11 и NBS1 к множественным сайтам повреждения ДНК» . Журнал биологической химии . 283 (2): 1197–208. дои : 10.1074/jbc.M706734200 . ПМИД 18025084 .
- ^ Jump up to: а б с Рогаков Е.П., Пильч Д.Р., Орр А.Х., Иванова В.С., Боннер В.М. (март 1998 г.). «Двухцепочечные разрывы ДНК индуцируют фосфорилирование гистона H2AX по серину 139» . Журнал биологической химии . 273 (10): 5858–68. дои : 10.1074/jbc.273.10.5858 . ПМИД 9488723 .
- ^ Мейланд Н., Беккер-Йенсен С., Фауструп Х., Меландер Ф., Бартек Дж., Лукас С. и др. (ноябрь 2007 г.). «RNF8 убиквитилирует гистоны в местах двухцепочечных разрывов ДНК и способствует сборке репарационных белков» . Клетка . 131 (5): 887–900. дои : 10.1016/j.cell.2007.09.040 . ПМИД 18001824 . S2CID 14232192 .
- ^ Луистербург М.С., Акс К., Акерманн Л., Вигант В.В., Беккер-Йенсен С., Ларсен Д.Х. и др. (май 2012 г.). «Новая некаталитическая роль убиквитинлигазы RNF8 в разворачивании структуры хроматина высшего порядка» . Журнал ЭМБО . 31 (11): 2511–27. дои : 10.1038/emboj.2012.104 . ПМЦ 3365417 . ПМИД 22531782 .
- ^ Jump up to: а б Луистербург М.С., Гоедхарт Дж., Мозер Дж., Кул Х., Гевертс Б., Хаутсмюллер А.Б. и др. (август 2007 г.). «Динамическое in vivo взаимодействие убиквитинлигазы DDB2 E3 с поврежденной УФ-излучением ДНК не зависит от белка распознавания повреждений XPC» . Журнал клеточной науки . 120 (Часть 15): 2706–16. дои : 10.1242/jcs.008367 . ПМИД 17635991 .
- ^ Jump up to: а б Пайнс А., Вруве М.Г., Мартейн Дж.А., Типас Д., Луистербург М.С., Кансой М. и др. (октябрь 2012 г.). «PARP1 способствует эксцизионному восстановлению нуклеотидов посредством стабилизации DDB2 и рекрутирования ALC1» . Журнал клеточной биологии . 199 (2): 235–49. дои : 10.1083/jcb.201112132 . ПМЦ 3471223 . ПМИД 23045548 .
- ^ Джазайери А., Фальк Дж., Лукас С., Бартек Дж., Смит Г.К., Лукас Дж. и др. (январь 2006 г.). «АТМ-зависимая от клеточного цикла регуляция ATR в ответ на двухцепочечные разрывы ДНК». Природная клеточная биология . 8 (1): 37–45. дои : 10.1038/ncb1337 . ПМИД 16327781 . S2CID 9797133 .
- ^ Баккенист CJ, Кастан М.Б. (январь 2003 г.). «Повреждение ДНК активирует АТМ посредством межмолекулярного аутофосфорилирования и диссоциации димеров». Природа . 421 (6922): 499–506. Бибкод : 2003Natur.421..499B . дои : 10.1038/nature01368 . PMID 12556884 . S2CID 4403303 .
- ^ Вэй Ц, Ли Л, Чен Д (2007). Восстановление ДНК, генетическая нестабильность и рак . Всемирная научная. ISBN 978-981-270-014-8 . [ нужна страница ]
- ^ Шонталь А.Х. (2004). Контроль контрольно-пропускных пунктов и рак . Хумана Пресс. ISBN 978-1-58829-500-2 . [ нужна страница ]
- ^ Гартель А.Л., Тайнер А.Л. (июнь 2002 г.). «Роль ингибитора циклинзависимой киназы p21 в апоптозе» . Молекулярная терапия рака . 1 (8): 639–49. ПМИД 12479224 .
- ^ Джанион С (2001). «Некоторые аспекты системы реагирования SOS — критический обзор» . Акта Биохимика Полоника . 48 (3): 599–610. дои : 10.18388/abp.2001_3894 . ПМИД 11833768 .
- ^ Jump up to: а б Эрилл И., Кампой С., Барбе Дж. (ноябрь 2007 г.). «Эоны страданий: эволюционный взгляд на бактериальную реакцию SOS» . Обзоры микробиологии FEMS . 31 (6): 637–56. дои : 10.1111/j.1574-6976.2007.00082.x . ПМИД 17883408 .
- ^ Шлахер К., Фам П., Кокс М.М., Гудман М.Ф. (февраль 2006 г.). «Роль ДНК-полимеразы V и белка RecA в мутации, вызванной SOS-повреждением». Химические обзоры . 106 (2): 406–19. дои : 10.1021/cr0404951 . ПМИД 16464012 .
- ^ Фрай Р.К., Бегли Т.Дж., Самсон Л.Д. (2004). «Общегеномные реакции на агенты, повреждающие ДНК». Ежегодный обзор микробиологии . 59 : 357–77. дои : 10.1146/annurev.micro.59.031805.133658 . ПМИД 16153173 .
- ^ Эспехель С., Мартин М., Клатт П., Мартин-Кабальеро Х., Флорес Х.М., Бласко М.А. (май 2004 г.). «Более короткие теломеры, ускоренное старение и увеличение лимфомы у мышей с дефицитом ДНК-PKcs» . Отчеты ЭМБО . 5 (5): 503–09. дои : 10.1038/sj.embor.7400127 . ПМК 1299048 . ПМИД 15105825 .
- ^ де Бур Дж., Андрессу Дж.О., де Вит Дж., Хейманс Дж., Бимс Р.Б., ван Стег Х. и др. (май 2002 г.). «Преждевременное старение у мышей с дефицитом репарации и транскрипции ДНК» . Наука . 296 (5571): 1276–79. Бибкод : 2002Sci...296.1276D . дои : 10.1126/science.1070174 . ПМИД 11950998 . S2CID 41930529 .
- ^ Долле М.Э., Бусуттил Р.А., Гарсия А.М., Вейнховен С., ван Друнен Э., Нидернхофер Л.Дж. и др. (апрель 2006 г.). «Повышенная геномная нестабильность не является предпосылкой для сокращения продолжительности жизни мышей с дефицитом репарации ДНК». Мутационные исследования . 596 (1–2): 22–35. Бибкод : 2006MRFMM.596...22D . дои : 10.1016/j.mrfmmm.2005.11.008 . ПМИД 16472827 .
- ^ Jump up to: а б с Макрей С.Л., Крокен М.М., Колдер Р.Б., Алипер А., Милхолланд Б., Уайт Р.Р., Жаворонков А., Гладышев В.Н., Селуанов А., Горбунова В., Чжан З.Д., Вийг Дж. (2015). «Репарация ДНК у видов с резкими различиями в продолжительности жизни». Старение. 7 (12): 1171–84. doi:10.18632/aging.100866. ПМЦ 4712340. ПМИД 26729707
- ^ Кобаяши Ю., Наруми И., Сато К., Фунаяма Т., Кикучи М., Китаяма С. и др. (ноябрь 2004 г.). «Механизмы радиационной реакции чрезвычайно радиорезистентной бактерии Deinococcus radiodurans». Маленький Сейбуцу Кагаку 18 (3): 134–35. ПМИД 15858357 .
- ^ Шпиндлер С.Р. (сентябрь 2005 г.). «Быстрая и обратимая индукция долголетия, противораковые и геномные эффекты ограничения калорий». Механизмы старения и развития . 126 (9): 960–66. дои : 10.1016/j.mad.2005.03.016 . ПМИД 15927235 . S2CID 7067036 .
- ^ Халика Х.Д., Чжао Х., Ли Дж., Ли Ю.С., Се Т.С., Ву Дж.М. и др. (декабрь 2012 г.). «Потенциальные антивозрастные агенты подавляют уровень конститутивной передачи сигналов mTOR и повреждения ДНК» . Старение . 4 (12): 952–65. дои : 10.18632/aging.100521 . ПМЦ 3615161 . ПМИД 23363784 .
- ^ Тиссенбаум Х.А., Гуаренте Л. (март 2001 г.). «Увеличенная дозировка гена sir-2 продлевает продолжительность жизни Caenorhabditis elegans». Природа . 410 (6825): 227–30. Бибкод : 2001Natur.410..227T . дои : 10.1038/35065638 . ПМИД 11242085 . S2CID 4356885 .
- ^ Коэн Х.И., Миллер С., Биттерман К.Дж., Уолл Н.Р., Хеккинг Б., Кесслер Б. и др. (июль 2004 г.). «Ограничение калорий способствует выживанию клеток млекопитающих, индуцируя деацетилазу SIRT1». Наука . 305 (5682): 390–92. Бибкод : 2004Sci...305..390C . дои : 10.1126/science.1099196 . ПМИД 15205477 . S2CID 33503081 .
- ^ Кабелоф, округ Колумбия, Янамадала С., Раффул Дж.Дж., Го З., Суфи А., Хейдари А.Р. (март 2003 г.). «Ограничение калорий способствует стабильности генома за счет индукции эксцизионной репарации основания и обращения вспять ее возрастного снижения». Восстановление ДНК . 2 (3): 295–307. дои : 10.1016/S1568-7864(02)00219-7 . ПМИД 12547392 .
- ^ Стюарт Дж.А., Карахалил Б., Хог Б.А., Соуза-Пинто Н.К., Бор В.А. (март 2004 г.). «Ограничение калорий по-разному влияет на восстановление митохондриальных и ядерных оснований ДНК» . Журнал ФАСЭБ . 18 (3): 595–97. doi : 10.1096/fj.03-0890fje . ПМИД 14734635 . S2CID 43118901 .
- ^ Уокер Д.В., МакКолл Г., Дженкинс Н.Л., Харрис Дж., Литгоу Г.Дж. (май 2000 г.). «Эволюция продолжительности жизни C. elegans». Природа . 405 (6784): 296–97. дои : 10.1038/35012693 . ПМИД 10830948 . S2CID 4402039 .
- ^ Джонсон Дж. (28 декабря 2010 г.). «Раскопки доисторических опухолей и дебаты» . Нью-Йорк Таймс .
Если бы мы жили достаточно долго, рано или поздно мы все заболели бы раком.
- ^ Альбертс Б., Джонсон А., Льюис Дж. и др. (2002). «Предупредимые причины рака» . Молекулярная биология клетки (4-е изд.). Нью-Йорк: Garland Science. ISBN 978-0-8153-4072-0 .
Определенную неснижаемую фоновую заболеваемость раком следует ожидать независимо от обстоятельств: мутаций никогда нельзя полностью избежать, поскольку они являются неизбежным следствием фундаментальных ограничений точности репликации ДНК, как обсуждалось в главе 5. Если бы человек мог жить долго более того, неизбежно, что по крайней мере одна из его или ее клеток в конечном итоге накопит набор мутаций, достаточный для развития рака.
- ^ Фриденсон Б. (август 2007 г.). «Путь BRCA1/2 предотвращает гематологический рак в дополнение к раку молочной железы и яичников» . БМК Рак . 7 : 152. дои : 10.1186/1471-2407-7-152 . ЧВК 1959234 . ПМИД 17683622 .
- ^ Гаванде Н.С., ВандерВере-Кароцца П.С., Хиншоу Х.Д., Джалал С.И., Сирс С.Р., Павелчак К.С. и др. (апрель 2016 г.). «Таргетная терапия по восстановлению ДНК: прошлое или будущее лечения рака?» . Фармакология и терапия . 160 : 65–83. doi : 10.1016/j.pharmthera.2016.02.003 . ПМЦ 4811676 . ПМИД 26896565 .
- ^ Брайант Х.Э., Шульц Н., Томас Х.Д., Паркер К.М., Флауэр Д., Лопес Э. и др. (апрель 2005 г.). «Специфическое уничтожение опухолей с дефицитом BRCA2 ингибиторами поли(АДФ-рибозо)-полимеразы». Природа . 434 (7035): 913–17. Бибкод : 2005Natur.434..913B . дои : 10.1038/nature03443 . ПМИД 15829966 . S2CID 4391043 .
- ^ Гольдштейн М., Кастан М.Б. (2015). «Реакция на повреждение ДНК: последствия для реакции опухоли на радиацию и химиотерапию». Ежегодный обзор медицины . 66 : 129–43. doi : 10.1146/annurev-med-081313-121208 . ПМИД 25423595 .
- ^ Jump up to: а б Джегго П.А., Перл Л.Х., Карр А.М. (январь 2016 г.). «Восстановление ДНК, стабильность генома и рак: историческая перспектива» (PDF) . Обзоры природы. Рак . 16 (1): 35–42. дои : 10.1038/nrc.2015.4 . ПМИД 26667849 . S2CID 14941857 .
- ^ Барткова Дж., Хорейси З., Коед К., Кремер А., Торт Ф., Цигер К. и др. (апрель 2005 г.). «Реакция на повреждение ДНК как кандидатный противораковый барьер на раннем этапе онкогенеза человека». Природа . 434 (7035): 864–70. Бибкод : 2005Natur.434..864B . дои : 10.1038/nature03482 . ПМИД 15829956 . S2CID 4398393 .
- ^ Jump up to: а б Барткова Дж., Резаи Н., Лионтос М., Каракайдос П., Клетсас Д., Исаева Н. и др. (ноябрь 2006 г.). «Онкоген-индуцированное старение является частью барьера онкогенеза, создаваемого контрольными точками повреждения ДНК». Природа . 444 (7119): 633–37. Бибкод : 2006Natur.444..633B . дои : 10.1038/nature05268 . ПМИД 17136093 . S2CID 4406956 .
- ^ Гайяр Х., Гарсиа-Муза Т., Агилера А. (май 2015 г.). «Репликативный стресс и рак». Обзоры природы. Рак . 15 (5): 276–89. дои : 10.1038/nrc3916 . hdl : 10261/123721 . ПМИД 25907220 . S2CID 11342123 .
- ^ Халазонетис Т.Д., Горгулис В.Г., Бартек Дж. (март 2008 г.). «Модель повреждения ДНК, вызванного онкогеном, для развития рака». Наука . 319 (5868): 1352–55. Бибкод : 2008Sci...319.1352H . дои : 10.1126/science.1140735 . ПМИД 18323444 . S2CID 16426080 .
- ^ де Бур Дж., Хоймейкерс Дж. Х. (март 2000 г.). «Репарация эксцизией нуклеотидов и человеческие синдромы» (PDF) . Канцерогенез . 21 (3): 453–60. дои : 10.1093/carcin/21.3.453 . PMID 10688865 .
- ^ Брустас К.Г., Либерман Х.Б. (февраль 2014 г.). «Гены реакции на повреждение ДНК и развитие метастазов рака» . Радиационные исследования . 181 (2): 111–30. Бибкод : 2014РадР..181..111Б . дои : 10.1667/RR13515.1 . ПМК 4064942 . ПМИД 24397478 .
- ^ Чжан П., Ван Дж., Гао В., Юань Б.З., Роджерс Дж., Рид Э. (май 2004 г.). «Экспрессия киназы CHK2 снижается из-за метилирования промотора при немелкоклеточном раке легких» . Молекулярный рак . 3 (4): 14. дои : 10.1186/1476-4598-3-14 . ПМК 419366 . ПМИД 15125777 .
- ^ Бэйлин С.Б. , Ом Дж.Э. (февраль 2006 г.). «Эпигенетическое подавление генов при раке - механизм ранней зависимости от онкогенного пути?». Обзоры природы. Рак . 6 (2): 107–16. дои : 10.1038/nrc1799 . ПМИД 16491070 . S2CID 2514545 .
- ^ Канвал Р., Гупта С. (апрель 2012 г.). «Эпигенетические модификации рака» . Клиническая генетика . 81 (4): 303–11. дои : 10.1111/j.1399-0004.2011.01809.x . ПМК 3590802 . ПМИД 22082348 .
- ^ Бальдасарре Дж., Баттиста С., Беллетти Б., Такур С., Пентималли Ф., Трапассо Ф. и др. (апрель 2003 г.). «Негативная регуляция экспрессии гена BRCA1 белками HMGA1 объясняет снижение уровня белка BRCA1 при спорадической карциноме молочной железы» . Молекулярная и клеточная биология . 23 (7): 2225–38. дои : 10.1128/MCB.23.7.2225-2238.2003 . ПМК 150734 . ПМИД 12640109 .
- ^ Хасинто Ф.В., Эстеллер М. (июль 2007 г.). «Мутаторные пути, вызванные эпигенетическим молчанием при раке человека» . Мутагенез . 22 (4): 247–53. дои : 10.1093/mutage/gem009 . ПМИД 17412712 .
- ^ Латц К., Пфайфер GP (февраль 2011 г.). «Эпигенетические изменения генов репарации ДНК при раке» . Журнал молекулярно-клеточной биологии . 3 (1): 51–58. дои : 10.1093/jmcb/mjq053 . ПМК 3030973 . ПМИД 21278452 . Эпигенетические изменения генов репарации ДНК при раке
- ^ Бернштейн С., Нфонсам В., Прасад А.Р., Бернштейн Х. (март 2013 г.). «Дефекты эпигенетического поля при прогрессировании рака» . Всемирный журнал желудочно-кишечной онкологии . 5 (3): 43–49. дои : 10.4251/wjgo.v5.i3.43 . ПМЦ 3648662 . ПМИД 23671730 .
- ^ Нарайанан Л., Фритцелл Дж.А., Бейкер С.М., Лискай Р.М., Глейзер П.М. (апрель 1997 г.). «Повышенные уровни мутаций во многих тканях мышей с дефицитом гена репарации несоответствия ДНК Pms2» . Труды Национальной академии наук Соединенных Штатов Америки . 94 (7): 3122–27. Бибкод : 1997PNAS...94.3122N . дои : 10.1073/pnas.94.7.3122 . ЧВК 20332 . ПМИД 9096356 .
- ^ Хеган Д.С., Нараянан Л., Жирик Ф.Р., Эдельманн В., Лискай Р.М., Глейзер П.М. (декабрь 2006 г.). «Различные модели генетической нестабильности у мышей с дефицитом генов репарации ошибочного спаривания Pms2, Mlh1, Msh2, Msh3 и Msh6» . Канцерогенез . 27 (12): 2402–08. doi : 10.1093/carcin/bgl079 . ПМК 2612936 . ПМИД 16728433 .
- ^ Тутт А.Н., ван Остром КТ, Росс ГМ, ван Стиг Х., Эшворт А. (март 2002 г.). «Нарушение Brca2 увеличивает частоту спонтанных мутаций in vivo: синергизм с ионизирующим излучением» . Отчеты ЭМБО . 3 (3): 255–60. дои : 10.1093/embo-reports/kvf037 . ПМК 1084010 . ПМИД 11850397 .
- ^ Герман Дж (март 1969 г.). «Синдром Блума. I. Генетические и клинические наблюдения у первых двадцати семи больных» . Американский журнал генетики человека . 21 (2): 196–227. ПМК 1706430 . ПМИД 5770175 .
- ^ Jump up to: а б с О'Хаган Х.М., Мохаммад Х.П., Бэйлин С.Б. (август 2008 г.). «Двухнитевые разрывы могут инициировать молчание генов и SIRT1-зависимое начало метилирования ДНК на экзогенном промоторном острове CpG» . ПЛОС Генетика . 4 (8): е1000155. дои : 10.1371/journal.pgen.1000155 . ПМЦ 2491723 . ПМИД 18704159 .
- ^ Куоццо С., Порчеллини А., Ангризано Т., Морано А., Ли Б., Ди Пардо А. и др. (июль 2007 г.). «Повреждение ДНК, репарация, направленная на гомологию, и метилирование ДНК» . ПЛОС Генетика . 3 (7): е110. дои : 10.1371/journal.pgen.0030110 . ЧВК 1913100 . ПМИД 17616978 .
- ^ Малкин Д. (апрель 2011 г.). «Синдром Ли-фраумени» . Гены и рак . 2 (4): 475–84. дои : 10.1177/1947601911413466 . ПМЦ 3135649 . ПМИД 21779515 .
- ^ Фирон Э.Р. (ноябрь 1997 г.). «Синдромы рака человека: ключ к разгадке происхождения и природы рака». Наука . 278 (5340): 1043–50. Бибкод : 1997Sci...278.1043F . дои : 10.1126/science.278.5340.1043 . ПМИД 9353177 .
- ^ Хэлфорд С., Роуэн А., Сойер Э., Талбот I, Томлинсон I (июнь 2005 г.). «О (6)-метилгуанин метилтрансфераза при колоректальном раке: обнаружение мутаций, потеря экспрессии и слабая связь с переходами G: C> A: T» . Гут . 54 (6): 797–802. дои : 10.1136/gut.2004.059535 . ПМК 1774551 . ПМИД 15888787 .
- ^ Шен Л., Кондо Ю., Рознер Г.Л., Сяо Л., Эрнандес Н.С., Вилайтонг Дж. и др. (сентябрь 2005 г.). «Метилирование промотора MGMT и дефект поля при спорадическом колоректальном раке» . Журнал Национального института рака . 97 (18): 1330–38. дои : 10.1093/jnci/dji275 . ПМИД 16174854 .
- ^ Псофаки В., Калогера С., Цамбурас Н., Стефану Д., Цианос Е., Сефериадис К. и др. (июль 2010 г.). «Статус метилирования промотора hMLH1, MGMT и CDKN2A/p16 при колоректальных аденомах» . Всемирный журнал гастроэнтерологии . 16 (28): 3553–60. дои : 10.3748/wjg.v16.i28.3553 . ПМК 2909555 . ПМИД 20653064 .
- ^ Ли К.Х., Ли Дж.С., Нам Дж.Х., Чой С., Ли MC, Парк К.С. и др. (октябрь 2011 г.). «Статус метилирования промотора генов hMLH1, hMSH2 и MGMT при колоректальном раке, связанном с последовательностью аденома-карцинома». Архив хирургии Лангенбека . 396 (7): 1017–26. дои : 10.1007/s00423-011-0812-9 . ПМИД 21706233 . S2CID 8069716 .
- ^ Амату А., Сарторе-Бьянки А., Моутинью С., Белотти А., Бенкардино К., Кирико Г. и др. (апрель 2013 г.). «Гиперметилирование промоторного CpG-островка фермента репарации ДНК MGMT предсказывает клинический ответ на дакарбазин в исследовании фазы II метастатического колоректального рака» . Клинические исследования рака . 19 (8): 2265–72. дои : 10.1158/1078-0432.CCR-12-3518 . ПМИД 23422094 .
- ^ Мокаррам П., Замани М., Кавусипур С., Нагибальхоссайни Ф., Ираджи С., Моради Сараби М. и др. (май 2013 г.). «Различные закономерности метилирования ДНК двух различных областей промотора O6-метилгуанин-ДНК-метилтрансферазы (O6-MGMT) при колоректальном раке». Отчеты по молекулярной биологии . 40 (5): 3851–57. дои : 10.1007/s11033-012-2465-3 . ПМИД 23271133 . S2CID 18733871 .
- ^ Трунингер К., Менигатти М., Луз Дж., Рассел А., Хайдер Р., Гебберс Дж.О. и др. (май 2005 г.). «Иммуногистохимический анализ выявляет высокую частоту дефектов PMS2 при колоректальном раке» . Гастроэнтерология . 128 (5): 1160–71. дои : 10.1053/j.gastro.2005.01.056 . ПМИД 15887099 .
- ^ Валери Н., Гаспарини П., Фаббри М., Бракони С., Веронезе А., Ловат Ф. и др. (апрель 2010 г.). «Модуляция репарации ошибочных спариваний и стабильности генома с помощью миР-155» . Труды Национальной академии наук Соединенных Штатов Америки . 107 (15): 6982–87. Бибкод : 2010PNAS..107.6982V . дои : 10.1073/pnas.1002472107 . ПМЦ 2872463 . ПМИД 20351277 .
- ^ Фациста А., Нгуен Х., Льюис С., Прасад А.Р., Рэмси Л., Зайтлин Б. и др. (апрель 2012 г.). «Недостаточная экспрессия ферментов репарации ДНК на ранней стадии развития спорадического рака толстой кишки» . Целостность генома . 3 (1): 3. дои : 10.1186/2041-9414-3-3 . ПМК 3351028 . ПМИД 22494821 .
- ↑ Гены восстановления ДНК человека , 15 апреля 2014 г., Онкологический центр доктора медицины Андерсона, Техасский университет.
- ^ Джин Б., Робертсон К.Д. (2013). «ДНК-метилтрансферазы, восстановление повреждений ДНК и рак». Эпигенетические изменения в онкогенезе . Достижения экспериментальной медицины и биологии. Том. 754. стр. 3–29. дои : 10.1007/978-1-4419-9967-2_1 . ISBN 978-1-4419-9966-5 . ПМК 3707278 . ПМИД 22956494 .
- ^ Кришнан К., Степто А.Л., Мартин Х.К., Вани С., Нонес К., Уодделл Н. и др. (февраль 2013 г.). «МикроРНК-182-5p нацелена на сеть генов, участвующих в репарации ДНК» . РНК . 19 (2): 230–42. дои : 10.1261/rna.034926.112 . ПМК 3543090 . ПМИД 23249749 .
- ^ Чайсаингмонгкол Дж., Попанда О., Варта Р., Дайкхофф Г., Херпель Е., Гейзельхарт Л. и др. (декабрь 2012 г.). «Эпигенетический скрининг генов репарации ДНК человека выявляет аберрантное метилирование промотора NEIL1 при плоскоклеточном раке головы и шеи» . Онкоген . 31 (49): 5108–16. дои : 10.1038/onc.2011.660 . ПМИД 22286769 .
- ^ Сингх П., Ян М., Дай Х., Ю Д., Хуан К., Тан В. и др. (ноябрь 2008 г.). «Сверхэкспрессия и гипометилирование гена лоскутной эндонуклеазы 1 при раке молочной железы и других видах рака» . Молекулярные исследования рака . 6 (11): 1710–17. дои : 10.1158/1541-7786.MCR-08-0269 . ПМЦ 2948671 . ПМИД 19010819 .
- ^ Лам Дж.С., Селигсон Д.Б., Ю Х., Ли А., Иева М., Пантак А.Дж. и др. (август 2006 г.). «Эндонуклеаза лоскута 1 сверхэкспрессируется при раке простаты и связана с высоким показателем Глисона». БЖУ Интернешнл . 98 (2): 445–51. дои : 10.1111/j.1464-410X.2006.06224.x . ПМИД 16879693 . S2CID 22165252 .
- ^ Ким Дж.М., Сон ХИ, Юн С.И., О Дж.Х., Ян Джо, Ким Дж.Х. и др. (январь 2005 г.). «Идентификация генов, связанных с раком желудка, с использованием микроматрицы кДНК, содержащей новые метки экспрессируемых последовательностей, экспрессируемых в клетках рака желудка» . Клинические исследования рака . 11 (2, часть 1): 473–82. дои : 10.1158/1078-0432.473.11.2 . ПМИД 15701830 .
- ^ Ван К., Се С., Чен Д. (май 2014 г.). «Эндонуклеаза лоскута 1 является многообещающим биомаркером-кандидатом при раке желудка и участвует в пролиферации и апоптозе клеток» . Международный журнал молекулярной медицины . 33 (5): 1268–74. дои : 10.3892/ijmm.2014.1682 . ПМИД 24590400 .
- ^ Краузе А., Комбаре В., Яконо И., Лакруа Б., Компаньон С., Бержерон С. и др. (июль 2005 г.). «Полногеномный анализ экспрессии генов в нейробластомах, обнаруженных с помощью массового скрининга» (PDF) . Раковые письма . 225 (1): 111–20. дои : 10.1016/j.canlet.2004.10.035 . ПМИД 15922863 . S2CID 44644467 .
- ^ Якобузио-Донахью К.А., Майтра А., Олсен М., Лоу А.В., ван Хик Н.Т., Рости С. и др. (апрель 2003 г.). «Исследование глобальных закономерностей экспрессии генов при аденокарциноме поджелудочной железы с использованием микрочипов кДНК» . Американский журнал патологии . 162 (4): 1151–62. дои : 10.1016/S0002-9440(10) 63911-9 ПМЦ 1851213 . ПМИД 12651607 .
- ^ Сато М., Жирар Л., Секине И., Сунага Н., Рамирес Р.Д., Камибаяши С. и др. (октябрь 2003 г.). «Повышенная экспрессия и отсутствие мутаций гена эндонуклеазы Flap (FEN1) при раке легких человека» . Онкоген . 22 (46): 7243–46. дои : 10.1038/sj.onc.1206977 . ПМИД 14562054 .
- ^ Би ФФ, Ли Д, Ян Кью (2013). «Гипометилирование сайтов связывания транскрипционных факторов ETS и усиление экспрессии PARP1 при раке эндометрия» . БиоМед Исследования Интернэшнл . 2013 : 946268. doi : 10.1155/2013/946268 . ПМЦ 3666359 . ПМИД 23762867 .
- ^ Би ФФ, Ли Д, Ян Ц (февраль 2013 г.). «Гипометилирование промотора, особенно вокруг мотива, специфичного для трансформации E26, и повышенная экспрессия поли(АДФ-рибозы) полимеразы 1 при серозном раке яичников с мутацией BRCA» . БМК Рак . 13:90 . дои : 10.1186/1471-2407-13-90 . ПМК 3599366 . ПМИД 23442605 .
- ^ Супек Ф., Ленер Б. (май 2015 г.). «Дифференциальное восстановление несоответствий ДНК лежит в основе изменения скорости мутаций в геноме человека» . Природа . 521 (7550): 81–84. Бибкод : 2015Natur.521...81S . дои : 10.1038/nature14173 . ПМЦ 4425546 . ПМИД 25707793 .
- ^ Jump up to: а б Чжэн К.Л., Ван Н.Дж., Чунг Дж., Мослехи Х., Санборн Дж.З., Хур Дж.С. и др. (ноябрь 2014 г.). «Транскрипция восстанавливает восстановление ДНК в гетерохроматине, определяя региональную частоту мутаций в раковых геномах» . Отчеты по ячейкам . 9 (4): 1228–34. дои : 10.1016/j.celrep.2014.10.031 . ПМК 4254608 . ПМИД 25456125 .
- ^ Ли Ф., Мао Г., Тонг Д., Хуан Дж., Гу Л., Ян В. и др. (апрель 2013 г.). «Гистоновая метка H3K36me3 регулирует восстановление несоответствия ДНК человека посредством взаимодействия с MutSα» . Клетка . 153 (3): 590–600. дои : 10.1016/j.cell.2013.03.025 . ПМЦ 3641580 . ПМИД 23622243 .
- ^ Супек Ф., Ленер Б. (июль 2017 г.). «Сигнатуры кластерных мутаций показывают, что склонная к ошибкам репарация ДНК нацелена на мутации в активных генах» . Клетка . 170 (3): 534–547.e23. дои : 10.1016/j.cell.2017.07.003 . hdl : 10230/35343 . ПМИД 28753428 .
- ^ Полак П., Лоуренс М.С., Хауген Э., Столецкий Н., Стоянов П., Турман Р.Э. и др. (январь 2014 г.). «Снижение плотности локальных мутаций в регуляторной ДНК раковых геномов связано с репарацией ДНК» . Природная биотехнология . 32 (1): 71–75. дои : 10.1038/nbt.2778 . ПМК 4116484 . ПМИД 24336318 .
- ^ Свенберг Дж.А., Лу К., Мёллер Б.С., Гао Л., Аптон П.Б., Накамура Дж. и др. (март 2011 г.). «Эндогенные и экзогенные аддукты ДНК: их роль в канцерогенезе, эпидемиологии и оценке риска» . Токсикол . 120 (Приложение 1): С130–45. дои : 10.1093/toxsci/kfq371 . ПМК 3043087 . ПМИД 21163908 .
- ^ Jump up to: а б Гамильтон М.Л., Го З., Фуллер К.Д., Ван Реммен Х., Уорд В.Ф., Остад С.Н. и др. (май 2001 г.). «Надежная оценка уровней 8-оксо-2-дезоксигуанозина в ядерной и митохондриальной ДНК с использованием метода йодида натрия для выделения ДНК» . Нуклеиновые кислоты Рез . 29 (10): 2117–26. дои : 10.1093/нар/29.10.2117 . ПМК 55450 . ПМИД 11353081 .
- ^ Минг Икс, Материя Б, Сонг М, Велиат Э, Шэнли Р, Джонс Р и др. (март 2014 г.). «Картирование структурно определенных продуктов окисления гуанина вдоль дуплексов ДНК: влияние контекста локальной последовательности и эндогенного метилирования цитозина» . J Am Chem Soc . 136 (11): 4223–35. дои : 10.1021/ja411636j . ПМЦ 3985951 . ПМИД 24571128 .
- ^ Jump up to: а б с Чжоу X, Чжуан Z, Ван В, Хэ Л, Ву Х, Цао Ю и др. (сентябрь 2016 г.). «OGG1 необходим для деметилирования ДНК, вызванного окислительным стрессом». Сигнал ячейки . 28 (9): 1163–1171. doi : 10.1016/j.cellsig.2016.05.021 . ПМИД 27251462 .
- ^ Поетч А.Р. (2020). «Геномика окислительного повреждения ДНК, репарации и последующего мутагенеза» . Comput Struct Biotechnol Дж . 18 : 207–219. дои : 10.1016/j.csbj.2019.12.013 . ПМК 6974700 . ПМИД 31993111 .
- ^ Д'Огюстен О, Уэ С., Кампаланс А, Радичелла Дж. П. (ноябрь 2020 г.). «Затерянный в толпе: как человеческая 8-оксогуанин ДНК-гликозилаза 1 (OGG1) находит 8-оксогуанин в геноме?» . Int J Mol Sci . 21 (21): 8360. doi : 10.3390/ijms21218360 . ПМЦ 7664663 . ПМИД 33171795 .
- ^ Лан Л., Накадзима С., Оохата Ю., Такао М., Окано С., Масутани М. и др. (сентябрь 2004 г.). «Анализ in situ процессов репарации окислительных повреждений ДНК в клетках млекопитающих» . Proc Natl Acad Sci США . 101 (38): 13738–43. Бибкод : 2004PNAS..10113738L . дои : 10.1073/pnas.0406048101 . ПМК 518826 . ПМИД 15365186 .
- ^ Чжоу X, Чжуан Z, Ван В, Хэ Л, Ву Х, Цао Ю и др. (сентябрь 2016 г.). «OGG1 необходим для деметилирования ДНК, вызванного окислительным стрессом». Сигнал ячейки . 28 (9): 1163–1171. doi : 10.1016/j.cellsig.2016.05.021 . ПМИД 27251462 .
- ^ Мур Л.Д., Ле Т., Фан Дж. (январь 2013 г.). «Метилирование ДНК и его основная функция» . Нейропсихофармакология . 38 (1): 23–38. дои : 10.1038/нпп.2012.112 . ПМК 3521964 . ПМИД 22781841 .
- ^ Маедер М.Л., Ангстман Дж.Ф., Ричардсон М.Е., Линдер С.Дж., Касио В.М., Цай С.К. и др. (декабрь 2013 г.). «Направленное деметилирование ДНК и активация эндогенных генов с использованием программируемых слитых белков TALE-TET1» . Нат. Биотехнология . 31 (12): 1137–42. дои : 10.1038/nbt.2726 . ПМЦ 3858462 . ПМИД 24108092 .
- ^ Дин Н., Бонэм Э.М., Хэннон Б.Е., Амик Т.Р., Бэйлин С.Б., О'Хаган Х.М. (июнь 2016 г.). «Белки репарации несоответствия привлекают ДНК-метилтрансферазу 1 к участкам окислительного повреждения ДНК» . J Мол Клеточная Биол . 8 (3): 244–54. дои : 10.1093/jmcb/mjv050 . ПМЦ 4937888 . ПМИД 26186941 .
- ^ Jump up to: а б Цзян З., Лай Ю., Бивер Дж.М., Цегай П.С., Чжао М.Л., Хортон Дж.К. и др. (январь 2020 г.). «Окислительное повреждение ДНК модулирует характер метилирования ДНК в гене рака молочной железы человека 1 (BRCA1) посредством перекрестных помех между ДНК-полимеразой β и ДНК-метилтрансферазой de novo» . Клетки . 9 (1): 225. doi : 10,3390/cells9010225 . ПМК 7016758 . ПМИД 31963223 .
- ^ Мортусевич О., Шермелле Л., Уолтер Дж., Кардосо М.К., Леонхардт Х. (июнь 2005 г.). «Привлечение ДНК-метилтрансферазы I к сайтам репарации ДНК» . Proc Natl Acad Sci США . 102 (25): 8905–9. Бибкод : 2005PNAS..102.8905M . дои : 10.1073/pnas.0501034102 . ПМЦ 1157029 . ПМИД 15956212 .
- ^ Jump up to: а б Куоццо С., Порчеллини А., Ангризано Т., Морано А., Ли Б., Ди Пардо А. и др. (июль 2007 г.). «Повреждение ДНК, репарация, направленная на гомологию, и метилирование ДНК» . ПЛОС Генет . 3 (7): е110. дои : 10.1371/journal.pgen.0030110 . ЧВК 1913100 . ПМИД 17616978 .
- ^ Ха К., Ли Г.Э., Палий С.С., Браун К.Д., Такеда Ю., Лю К. и др. (январь 2011 г.). «Быстрое и временное привлечение DNMT1 к двухцепочечным разрывам ДНК опосредовано его взаимодействием с множеством компонентов механизма реакции на повреждение ДНК» . Хум Мол Жене . 20 (1): 126–40. дои : 10.1093/hmg/ddq451 . ПМК 3000680 . ПМИД 20940144 .
- ^ Руссо Г., Ланди Р., Пезоне А., Морано А., Зученья С., Романо А. и др. (сентябрь 2016 г.). «Повреждение и восстановление ДНК изменяют метилирование ДНК и домен хроматина целевого локуса: механизм полиморфизма метилирования аллелей» . Научный представитель . 6 : 33222. Бибкод : 2016NatSR...633222R . дои : 10.1038/srep33222 . ПМК 5024116 . ПМИД 27629060 .
- ^ Фаррис М.Х., Текстер П.А., Мора А.А., Уайлс М.В., Мак Гарригл Э.Ф., Клаус С.А. и др. (декабрь 2020 г.). «Обнаружение CRISPR-опосредованных модификаций генома посредством изменения паттернов метилирования CpG-островков» . БМК Геномика . 21 (1): 856. doi : 10.1186/s12864-020-07233-2 . ПМК 7709351 . ПМИД 33267773 .
- ^ Аллен Б., Пезоне А., Порчеллини А., Мюллер М.Т., Мастернак М.М. (июнь 2017 г.). «Номологичные соединения концов вызывают изменения в метилировании ДНК: источник постоянных эпигенетических изменений» . Онкотаргет . 8 (25): 40359–40372. дои : 10.18632/oncotarget.16122 . ПМЦ 5522286 . ПМИД 28423717 .
- ^ Кроми Г.А., Коннелли Дж.К., Лич Д.Р. (декабрь 2001 г.). «Рекомбинация при двухцепочечных разрывах и концах ДНК: консервативные механизмы от фага до человека» . Молекулярная клетка . 8 (6): 1163–74. дои : 10.1016/S1097-2765(01)00419-1 . ПМИД 11779493 .
- ^ О'Брайен П.Дж. (февраль 2006 г.). «Каталитическая распущенность и расходящаяся эволюция ферментов репарации ДНК». Химические обзоры . 106 (2): 720–52. дои : 10.1021/cr040481v . ПМИД 16464022 .
- ^ Мареска Б., Шварц Дж. Х. (январь 2006 г.). «Внезапное происхождение: общий механизм эволюции, основанный на концентрации стрессовых белков и быстром изменении окружающей среды» . Анатомические записи, часть B: Новый анатом . 289 (1): 38–46. дои : 10.1002/ar.b.20089 . ПМИД 16437551 .
- ^ «Инструмент редактирования генов CRISPR привел ученых в восторг, но нервничает» Новости CBC. Автор Келли Кроу. 30 ноября 2015 г.
Внешние ссылки
[ редактировать ] СМИ, связанные с восстановлением ДНК, на Викискладе?
СМИ, связанные с восстановлением ДНК, на Викискладе? - Лекции по восстановлению ДНК Института рака Розуэлл-Парк
- Полный список генов восстановления ДНК человека
- 3D-структуры некоторых ферментов репарации ДНК
- Мачадо ЧР, Менк КФ (декабрь 1997 г.). «Болезни восстановления ДНК человека: от нестабильности генома к раку» . Браз. Ж. Жене . 20 (4): 755–762. дои : 10.1590/S0100-84551997000400032 .
- Группа специальных интересов по восстановлению ДНК
- Восстановление ДНК. Архивировано 12 февраля 2018 г. в Wayback Machine.
- Повреждение ДНК и восстановление ДНК
- Сегментарная прогерия
- Хакем Р. (февраль 2008 г.). «Восстановление повреждений ДНК: хорошее, плохое и ужасное» . ЭМБО Дж . 27 (4): 589–605. дои : 10.1038/emboj.2008.15 . ПМК 2262034 . ПМИД 18285820 .
- Моралес М.Э., Дербес Р.С., Аде К.М., Ортего Дж.К., Старк Дж., Дейнингер П.Л. и др. (2016). «Воздействие тяжелых металлов влияет на результаты восстановления двойной цепи ДНК» . ПЛОС ОДИН . 11 (3): e0151367. Бибкод : 2016PLoSO..1151367M . дои : 10.1371/journal.pone.0151367 . ПМЦ 4788447 . ПМИД 26966913 .
| Эксцизионное восстановление | |
|---|---|
| Гомологичная рекомбинация | |
| Другие пути | |
| Регулирование | |
| Другое/несгруппировано | |