Митохондриальная миопатия
| Митохондриальная миопатия | |
|---|---|
| Другие имена | Митохондриальные мышечные заболевания; мышечная митохондринопатия; мышечная митохондриальная дисфункция |
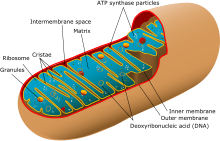 | |
| Упрощенная структура типичной митохондрии. | |
| Специальность | Нервно-мышечная медицина |
Митохондриальные миопатии — это типы миопатий, связанные с митохондриальными заболеваниями . [ 1 ] Аденозинтрифосфат ( АТФ ), химическое вещество, используемое для обеспечения клеток энергией, не может производиться в достаточной степени путем окислительного фосфорилирования , когда митохондрии либо повреждены, либо отсутствуют необходимые ферменты или транспортные белки. При недостаточном производстве АТФ в митохондриях возникает чрезмерная зависимость от анаэробного гликолиза, что приводит к лактоацидозу как в состоянии покоя, так и при физической нагрузке. [ 2 ]
Первичные митохондриальные миопатии передаются по наследству, тогда как вторичные митохондриальные миопатии могут передаваться по наследству (например, мышечная дистрофия Дюшенна). [ 3 ] или окружающей среды (например, алкогольная миопатия [ 4 ] [ 5 ] ). Когда это наследственное первичное заболевание, это одна из метаболических миопатий . [ 6 ] [ 4 ]
При биопсии мышечной ткани пациентов с этими заболеваниями обычно наблюдаются «рваные красные» мышечные волокна при окрашивании трихромом Гомори. Рвано-красный вид обусловлен скоплением аномальных митохондрий под плазматической мембраной. [ 7 ] Эти рваные красные волокна могут содержать нормальные или аномально повышенные скопления гликогена и нейтральных липидов, при этом гистохимическое окрашивание указывает на аномальное поражение дыхательной цепи, такое как снижение сукцинатдегидрогеназы или цитохром с-оксидазы . [ 8 ] Наследование считалось материнским ( неменделевское внеядерное ). Теперь известно, что определенные делеции ядерной ДНК также могут вызывать митохондриальную миопатию, например делецию гена OPA1 . [ 6 ]
Признаки и симптомы
[ редактировать ]Распространенными симптомами являются слабость проксимальных мышц, непереносимость физической нагрузки, лактоацидоз, высокое соотношение лактат/пируват в сыворотке, нормальный или повышенный уровень КФК в сыворотке, одышка, усиленная кардиореспираторная реакция на физическую нагрузку. Он может быть изолирован в мышцах (чистая миопатия) или может быть системным, включая не только миопатию, но также аномалии глаз, периферическую невропатию и неврологические нарушения. При биопсии мышц обычно обнаруживаются рваные красные волокна, гистохимическое окрашивание показывает аномалии дыхательной цепи или снижение цитохром-с-оксидазы (ЦОГ). [ 9 ] [ 10 ]
Пять наиболее распространенных — это MELAS, MERF, KSS, CPEO и MNGIE, которые перечислены ниже: [ 9 ]
- Митохондриальная энцефаломиопатия, лактоацидоз и инсультоподобный синдром (MELAS)
- Различные степени когнитивных нарушений и деменции
- Лактацидоз
- Штрихи
- Транзиторные ишемические атаки
- Потеря слуха
- Потеря веса
- Миоклоническая эпилепсия и рваные красные волокна (MERRF)
- Прогрессирующая миоклоническая эпилепсия
- Сгустки больных митохондрий накапливаются в мышечных волокнах и выглядят как «рваные красные волокна» при окрашивании мышц модифицированным трихромным красителем Гёмёри.
- Маленький рост
- Синдром Кернса-Сейра (КСС)
- Наружная офтальмоплегия
- Нарушения сердечной проводимости
- Нейросенсорная потеря слуха
- Хроническая прогрессирующая наружная офтальмоплегия (ХПЭО)
- Прогрессирующий офтальмопарез
- Симптоматика совпадает с другими митохондриальными миопатиями.
- Митохондриальная нейрогастроинтестинальная энцефалопатия (MNGIE)
- Мышечная слабость и атрофия, более выраженные в дистальной части.
- Гипорефлексный или арефлексический
- Частые птоз и офтальмопарез
- Нарушения моторики желудочно-кишечного тракта (например, вздутие живота, спазмы желудка, диарея)
Причина
[ редактировать ]
Митохондриальная миопатия буквально означает митохондриальное заболевание мышц, заболевание мышц, вызванное митохондриальной дисфункцией. Митохондрии являются основным производителем энергии почти во всех клетках организма. Исключением являются зрелые эритроциты (эритроциты), поэтому они не расходуют кислород, который они несут. В глазу хрусталик и наружный сегмент сетчатки почти не содержат митохондрий. Мышечные клетки имеют много митохондрий, особенно мышечных волокон типа I , и если у митохондрий возникают проблемы, из-за которых они не производят достаточно энергии для функционирования клетки, возникают проблемы. [ 11 ]
Причина может быть генетической: многие из них имеют митохондриальное наследование (включая митохондриальную ДНК, которая передается только от матери), хотя также существуют мутации ядерной ДНК с менделевским наследованием , которые являются либо аутосомно-доминантными, рецессивными, либо Х-сцепленными рецессивными. Примером ядерной ДНК является мутация в гене POLG (гамма-полимераза), которая приводит митохондриальной ДНК к повреждению и потере функции (мтДНК).
Список болезней
[ редактировать ]| Имя
(альтернативные названия) |
Ген(ы) | Шаблон наследования
(MT, AR, AD, X-Linked) |
ПРОПУСКАТЬ #
(GD: описание гена, PS: фенотипический ряд) |
|---|---|---|---|
| Митохондриальная энцефаломиопатия, лактоацидоз и инсультоподобный синдром (MELAS)
(Ювенильная миопатия, энцефалопатия, лактоацидоз и инсульт) |
MT-TL1 , MT-TQ , MT-TH , MT-TK , MT-TC , MT-TS1 , MT-ND1 , MT-ND5 , MT-ND6 , MT-TS2 | МТ | 540000 [ 12 ] |
| Миоклоническая эпилепсия и рваные красные волокна (MERRF) | МТ-ТК , МТ-ТЛ1 , МТ-ТХ , МТ-ТС1 , МТ-ТС2 , МТ-ТФ | МТ | 545000 [ 13 ] |
| Синдром Кернса-Сейра (КСС)
(Офтальмоплегия, пигментная дегенерация сетчатки и кардиомиопатия; окулокраниосоматический синдром; синдром офтальмоплегия-плюс; митохондриальная цитопатия, офтальмоплегия, прогрессирующая наружная, с рваными красными волокнами; хроническая прогрессирующая наружная офтальмоплегия с миопатией; CPEO с миопатией; CPEO с рваными красными волокнами). ) |
МТ-ТЛ1 | МТ | 530000 [ 14 ] |
| Хроническая прогрессирующая наружная офтальмоплегия (ХПЭО)
(Прогрессирующая наружная офтальмоплегия с делециями митохондриальной ДНК, аутосомно-рецессивный/доминантный тип) |
ПОЛГ , SLC25A4 , RNASEH1 , TWNK , TK2 , POLG2 , DGUOK , TOP3A , RRM2B | АР/АД | ПС157640 [ 15 ] |
| Синдром истощения митохондриальной ДНК (тип MNGIE)
(Митохондриальная нейрогастроинтестинальная энцефалопатия (MNGIE); синдром мионейро-гастроинтестинальной энцефалопатии; полинейропатия, офтальмоплегия, лейкоэнцефалопатия и псевдообструкция кишечника; синдром POLIP) |
ТИМП , РРМ2Б , ПОЛГ , ЛИГ3 | АР | 603041;
612075; 613662; 619780 [ 16 ] |
| Синдром истощения митохондриальной ДНК
(тип Альперса, кардиомиопатический тип, энцефаломиопатический тип, гепатоцеребральный тип и миопатический тип) |
MGME1 , SLC25A10 , TK2 , POLG , SLC25A21 , SUCLA2 , TWNK , TFAM , AGK , MRM2 , SLC25A4 , OPA1 , SUCLG1 | АР/АД | PS603041 [ 16 ] |
| Митохондриальная миопатия, инфантильная, транзиторная (ММИТ)
(Митохондриальная миопатия, инфантильная, преходящая, вследствие недостаточности дыхательной цепи; миопатия с дефицитом ЦОГ, инфантильная, преходящая; недостаточность дыхательной цепи, инфантильная, преходящая) |
МТ-ТЭ | МТ | 500009 [ 17 ] |
| Митохондриальная миопатия, летальная, инфантильная (LIMM)
(Летальная инфантильная митохондриальная миопатия) |
МТ-ТТ | МТ | 551000 [ 18 ] |
| Наследственная миопатия с лактоацидозом (HML)
(миопатия с непереносимостью физической нагрузки, шведский тип; миопатия с дефицитом сукцинатдегидрогеназы и аконитазы; миоглобинурия вследствие аномального гликолиза; синдром Ларссона-Линдерхольма; миопатия Линдерхольма) |
ВНЕ | АР/АД [ 19 ] | 255125 [ 20 ] |
| Митохондриальная миопатия при сахарном диабете
(Митохондриальная миопатия липидного типа) |
МТ-ТЭ | МТ | 500002 [ 21 ] |
| Наследственный диабет и глухота по материнской линии (MIDD)
(Диабет и глухота (DAD); синдром Баллинджера-Уоллеса; инсулиннезависимый сахарный диабет с глухотой, наследуемый по материнской линии) |
МТ-ТЛ1 , МТ-ТЕ , МТ-ТК | МТ | 520000 [ 22 ] |
| Миопатия митохондриальная прогрессирующая с врожденной катарактой и задержкой развития (MPMCD)
(Миопатия с катарактой и комбинированной недостаточностью дыхательной цепи; комбинированная недостаточность митохондриального комплекса) |
ВФЭР | АР | 613076 [ 23 ] |
| Миопатия, лактоацидоз и сидеробластная анемия (MLASA)
(Митохондриальная миопатия и сидеробластная анемия) |
ПУС1 , ЯРС2, МТ-АТП6 | АР/МТ | 600462
613561 [ 24 ] ГД: 516060 [ 25 ] |
| Миопатия, изолированная митохондриальная, аутосомно-доминантная (IMMD) | CHCHD10 | ОБЪЯВЛЕНИЕ | 616209 [ 26 ] |
| Миопатия, митохондриальная и атаксия (MMYAT) | МСТО1 | АР/АД | 617675 [ 27 ] |
| Митохондриальная миопатия, эпизодическая, с атрофией зрительного нерва или без нее и обратимая лейкоэнцефалопатия (МЕОАЛ) | FDX2 | АР | 251900 [ 28 ] |
| Митохондриальная миопатия с лактоацидозом (MMLA) | ПНПЛА8 | АР | 251950 [ 29 ] |
| Митохондриальная миопатия с дефектом транспорта митохондриальных белков | Неизвестный | АР | 251945 [ 30 ] |
| миотоническая дистрофиеподобная миопатия;
Митохондриальная миопатия |
МТ-ТА | МТ | Год: 590000 [ 31 ] |
| Митохондриальная миопатия, изолированная | МТ-ТД | МТ | ГД: 590015 [ 32 ] |
| Миопатия, митохондриальная | МТ-ТВ | МТ | ГД: 590095 [ 33 ] |
| коэнзима Q 10 Первичный дефицит (COQ10D)
(дефицит CoQ10, первичный; дефицит убихинона; дефицит коэнзима Q; дефицит CoQ) |
COQ2 , PDSS1 , PDSS2 , ADCK3 , COQ9 , COQ4 , COQ7 , COQ5 | АР | ПС607426 [ 34 ] |
| Дефицит митохондриального комплекса I ядерного типа (MC1DN)
(НАДН:Q(1)-оксидоредуктазный дефицит; НАДН-коэнзим Q-редуктазный дефицит; митохондриальный НАДН-дегидрогеназный компонент комплекса I, дефицит) |
NDUFS2 , NDUFB3 , NDUFS1 , NDUFA10 , NDUFAF3 , TIMMDC1 , ACAD9 , NDUFS6 , NDUFS4 , NDUFAF2 , NDUFA2 , NDUFAF4 , DNAJC30 , NDUFAF6 , NDUFB9 , NDUFA8 , NDUFB8 , NDUFS3 , NDUFV1 , НДУФС8 . NDUFC2 , TMEM126B , FOXRED1 , NDUFA9 , NDUFA12 , NUBPL , NDUFAF1 , MTFMT , NDUFB10 , NDUFAF8 , NDUFV2 , NDUFS7 , NDUFA11 , NDUFB7 , NDUFA13 , NDUFAF5 , NDUFA6 , NDUFB11 , ФА1 | АР/XL/XLR | ПС252010 [ 35 ] |
| Дефицит митохондриального комплекса II ядерного типа (MC2DN)
(дефицит сукцинат-CoQ-редуктазы; дефицит сукцинатдегидрогеназы) |
SDHA , SDHAF1 , SDHD , SDHB | АР | ПС252011 [ 36 ] |
| цитохром b комплекса III (MTCYB);
Проявлять непереносимость; мультисистемное расстройство; кардиомиопатия, инфантильный гистиоцитоид; непереносимость физической нагрузки, кардиомиопатия и септооптическая дисплазия; паркинсонизм/синдром перекрытия MELAS |
МТ-КИБ | МТ | ГД: 516020 [ 37 ] |
| Дефицит митохондриального комплекса III ядерного типа (MC3DN) | BCS1L , TTC19 , UQCRQ , UQCRC2 , CYC1 , UQCC2 , LYRM7 , UQCC3 , UQCRFS1 | АР | ПС124000 [ 38 ] |
| Дефицит митохондриального комплекса IV ядерного типа (MC4DN)
( дефицит митохондриального комплекса IV; дефицит цитохром с-оксидазы; дефицит ЦОГ) |
SURF1 , SCO2 , COX10 , SCO1 , LRPPRC , COX15 , COX6B1 , TACO1, COX14 , COX20 , PET100 , COA6 , COA3 , COX8A , COX4I1 , APOPT1, COX6A2 , PET117 , COX5A , ЦОГФА4, ЦОГ16, ЦОГ11 | АР | ПС220110 [ 39 ] |
| Дефицит митохондриального комплекса V (АТФ-синтазы) , ядерный тип (MC5DN) | ATPAF2 , TMEM70 , ATP5E , ATP5F1A , ATP5F1D, ATP5MD , ATP5PO | АР/АД | ПС604273 [ 40 ] |
| Мышечная дистрофия конечностно-поясного типа, 1Н тип.
(По состоянию на 2017 год был исключен из LGMD за наличие гистохимических доказательств митохондриальной миопатии, но еще не получил новую номенклатуру) [ 41 ] [ 42 ] |
Хромосома 3 (3p23-p25), неизвестный ген | ОБЪЯВЛЕНИЕ | 613530 [ 43 ] |
Диагностика
[ редактировать ]
Биопсия мышц: обычно рваные красные волокна при окраске трихромом Гёмори , нормальное или избыточное накопление гликогена или липидов в этих рваных красных волокнах, гистохимическое окрашивание показывает нарушение дыхательной цепи, например, ЦОГ-негативные волокна. [ 6 ] [ 8 ] Некоторые митохондриальные миопатии ограничиваются проявлением заболевания только в скелетных мышцах, при этом фибробласты (по данным биопсии кожи) выглядят нормальными. [ 44 ] [ 19 ]
Анализы крови: соотношение лактат/пируват может быть повышенным или нормальным, креатинкиназа (КК) может быть повышенной или нормальной. [ 6 ] [ 2 ] Электролитная панель, анионная разница, глюкоза, витамин D, ТТГ , аутоантитела к HMGCR и AChR для исключения псевдометаболических миопатий. [ 6 ] [ 2 ]
Нагрузочный тест: усиленная кардиореспираторная реакция на физическую нагрузку (неадекватная реакция учащенного пульса на физическую нагрузку с одышкой [ тахикардия и одышка ]). [ 10 ]
Тесты ДНК: нейромышечные панели секвенирования всего экзома ( WES ) (которые проверяют только экзоны ) или секвенирование всего генома ( WGS ) для более сложных случаев (которые проверяют экзоны, интроны и митохондриальную ДНК ). Первоначально считалось, что интроны представляют собой «мусорную ДНК», однако некоторые интроны регулируют экспрессию экзонов. [ 45 ] [ 46 ] Например, при митохондриальной миопатии наследственной миопатии с лактоацидозом (ГМЛ) наиболее распространенной патогенной мутацией является интронная IVS5+382 G>C (rs767000507). [ 19 ]
Есть две группы ДНК, влияющие на митохондрии: митохондриальный геном (мтДНК) и ядерная ДНК. [ 6 ] При митохондриальных миопатиях, которые включают одну делецию мтДНК, ее можно обнаружить только в мтДНК, полученной из мышц, поэтому для анализа ДНК необходима биопсия пораженной мышцы, а не слюна или кровь. [ 6 ] [ 8 ] Даже у братьев и сестер с одной и той же наследственной мутацией были затронуты разные группы мышц, при этом непораженные ткани имели почти нормальный уровень мтДНК. [ 47 ] [ 48 ]
ЭМГ: может быть нормальной, миопатической или редко нейрогенной. [ 6 ]
Симптомы непереносимости физической нагрузки, аномальная мышечная утомляемость, миалгия (мышечная боль), аритмия, возможная фиксированная слабость проксимальных мышц, липидные отложения, возможные эпизоды рабдомиолиза, причем симптомы становятся очевидными или ухудшаются натощак, во время лихорадки, во время аэробных занятий низкой интенсивности. активности или после длительной активности – все это совпадает с симптомами другой метаболической миопатии – нарушения обмена жирных кислот . [ 6 ]
ДНК-тестирование помогает определить сходные по проявлению, но разные по биоэнергетическому происхождению метаболические миопатии. Если анализ ДНК не дал результатов, необходима биопсия мышц. [ 2 ] [ 6 ] [ 8 ]
Дифференциальный диагноз
[ редактировать ]Заболевания, имитирующие симптомы митохондриальной миопатии, включают электролитный дисбаланс, миастению, нарушения функции щитовидной железы, дефицит витамина D, иммуноопосредованную некротизирующую миопатию, псевдогипоксию , связанную с диабетом , и нарушения метаболизма жирных кислот. [ 6 ] [ 2 ] Гипоксия вследствие ишемии (недостаточный кровоток) также нарушает окислительное фосфорилирование, что можно наблюдать при перемежающейся хромоте, хронической венозной недостаточности и синдроме ущемления подколенной артерии. Если симптомы мышечной усталости улучшаются примерно через 10 минут аэробных упражнений низкой и умеренной интенсивности или примерно через 10 минут отдыха после аэробных упражнений, это может указывать на феномен второго дыхания, наблюдаемый при некоторых мышечных гликогенозах. [ 2 ]
Рваные красные волокна (митохондриальная аномалия) можно обнаружить при ряде миопатий, помимо наследственных первичных митохондриальных миопатий. [ 49 ] К ним относятся аксональная болезнь Шарко-Мари-Тута типов 2CC и 2EE, врожденный миастенический синдром типов 12 и 14, врожденная миопатия типов 10B и 22A и MYH7 -связанные миопатии, такие как дистальная миопатия Лэнга и миопатия накопления миозина. [ 49 ]
Вторичная митохондриальная миопатия может быть вызвана естественным старением. [ 50 ] [ 51 ] воспалительные миопатии, [ 50 ] и хроническое расстройство, связанное с употреблением алкоголя. [ 4 ] [ 5 ] Это также может быть связано с приемом некоторых препаратов, таких как статины, бупивакаин, противоэпилептические препараты (фенитоин, вальпроевая кислота и ламотриджин) и нуклеозидные ингибиторы обратной транскриптазы (противовирусные препараты), такие как зидовудин и клевудин. [ 52 ]
Некоторые метаболические миопатии затрагивают несколько биоэнергетических путей, например, множественный дефицит ацил-КоА-дегидрогеназы (MADD) , ранее известный как глутаровая ацидемия типа II (GA-II). Гены ETF , участвующие в MADD, ухудшают бета-окисление (метаболизм жирных кислот), ухудшают катаболизм аминокислот (метаболизм белков) и одновременно нарушают дыхательную цепь, не перенося электроны от восстановленного FAD. + /ФАДХ 2 . Нарушение белкового обмена приводит к накоплению глутаровой кислоты и других кислот. Метаболизм жирных кислот еще больше ухудшается, поскольку карнитин используется для детоксикации накопления глутаровой кислоты, вызывая вторичный дефицит карнитина . [ 53 ] [ 54 ] Хотя MADD влияет на несколько биоэнергетических путей, его классифицируют как нарушение метаболизма жирных кислот, поскольку именно этот биоэнергетический путь больше всего страдает от дефицита. Тем не менее, это важно отметить при дифференциальном диагнозе, поскольку симптомы не только совпадают с митохондриальными миопатиями, но и биопсия мышц у некоторых людей с MADD показывает ЦОГ-негативные волокна, нарушение дыхательной цепи и дефицит коэнзима Q 10 . [ 55 ] [ 56 ] Некоторые формы MADD хорошо реагируют на рибофлавин, известные как рибофлавин-чувствительный MADD (RR-MADD). [ 6 ]
Рибофлавин-зависимая непереносимость физической нагрузки (RREI), нарушение метаболизма жирных кислот, связанное с геном SLC25A32 , имеет симптомы, сходные с MADD: при биопсии мышц обнаруживаются рваные красные волокна и липидные отложения (в основном в волокнах I типа), мелкие волокна II типа и нарушение FAD-зависимая дыхательная цепь митохондрий. [ 57 ]
Болезнь Помпе (гликогенная болезнь II типа), другой тип метаболической миопатии, имеет вторичную митохондриальную дисфункцию, присутствующую как в более ранних формах (инфантильных и юношеских), так и в поздних формах у взрослых. [ 58 ]
Миопатии с участием гена DMD , такие как мышечная дистрофия Дюшенна и Беккера , имеют вторичную митохондриальную дисфункцию, нарушающую окислительное фосфорилирование. [ 3 ] [ 59 ] Механизмов, приводящих к этой митохондриальной дисфункции, много, и еще предстоит выяснить, какие митохондриальные изменения непосредственно связаны с заболеванием, а какие являются компенсаторными. [ 3 ] У трех неродственных мальчиков с мутацией в гене МДД наблюдались псевдометаболические проявления с симптомами непереносимости физической нагрузки, проявляющимися в виде вызванной физической нагрузкой миалгии, мышечной ригидности, миоглобинурии и рабдомиолиза. [ 60 ]
некоторые мышечные дистрофии конечностей Известно, что имеют вторичную митохондриальную дисфункцию, в том числе: LGMDR1, связанную с кальпаином 3 ( ранее LGMD 2A ), LGMDR2, связанную с дисферлином ( LGMD 2B ), LGMDR3, связанную с α-саркогликаном ( LGMDR 2D ), LGMDR5 γ-. связанный с саркогликанами ( LGMD 2C ) и LGMDR6 Связанный с δ-саркогликаном ( LGMD 2F ). [ 59 ] [ 61 ] А также миофибриллярная миопатия 8 (MFM8), связанная с PYROXD1, которая имеет медленно прогрессирующий фенотип конечностей и пояса у взрослых. [ 59 ] [ 62 ]
Уход
[ редактировать ]Хотя лекарства в настоящее время не существует, есть надежда на лечение этого класса наследственных заболеваний, поскольку испытания продолжаются.
Аэробные тренировки могут улучшить окислительную способность скелетных мышц, которые становятся аэробными . Дезоксинуклеозидмонофосфаты и дезоксинуклеотиды, принимаемые перорально, могут помочь при дефиците ТК2 (синдром истощения митохондриальной ДНК 2 миопатического типа). [ 6 ]
Может помочь избегание физически стрессовых ситуаций, которые истощают запасы гликогена, таких как голодание и упражнения на выносливость (которые основаны преимущественно на окислительном фосфорилировании). Диета с высоким содержанием углеводов/низким содержанием жиров/белков может помочь. [ 6 ]
См. также
[ редактировать ]Ссылки
[ редактировать ]- ^ «Информационная страница митохондриальной миопатии | Национальный институт неврологических расстройств и инсульта» . www.ninds.nih.gov . Проверено 28 февраля 2017 г.
- ^ Перейти обратно: а б с д и ж Бхаи С. (сентябрь 2021 г.). «Нейромышечные заметки: диагностика метаболических миопатий» . Практическая неврология .
- ^ Перейти обратно: а б с Хайдеманн А (июнь 2018 г.). «Метаболизм скелетных мышц при мышечной дистрофии Дюшенна и Беккера – последствия для терапии» . Питательные вещества . 10 (6): 796. дои : 10.3390/nu10060796 . ПМК 6024668 . ПМИД 29925809 .
- ^ Перейти обратно: а б с Саймон Л., Джолли С.Е., Молина П.Е. (2017). «Алкогольная миопатия: патофизиологические механизмы и клинические последствия» . Исследование алкоголя . 38 (2): 207–217. ПМЦ 5513686 . ПМИД 28988574 .
- ^ Перейти обратно: а б Сон Б.Дж., Акбар М., Абдельмегид М.А., Бён К., Ли Б., Юн С.К., Хардвик Дж.П. (01.01.2014). «Митохондриальная дисфункция и повреждение тканей алкоголем, жирами, безалкогольными веществами и патологическими состояниями посредством посттрансляционных модификаций белков» . Редокс-биология . 3 : 109–123. дои : 10.1016/j.redox.2014.10.004 . ПМЦ 4297931 . ПМИД 25465468 . S2CID 17113550 .
- ^ Перейти обратно: а б с д и ж г час я дж к л м н Уртисбереа Дж.А., Севера Г., Малфатти Э. (апрель 2023 г.). «Метаболические миопатии в эпоху секвенирования следующего поколения» . Гены . 14 5):954.doi : ( 10.3390/gen14050954 . ПМЦ 10217901 . ПМИД 37239314 .
- ^ «Рваные красные мышечные волокна — MedGen — NCBI» . www.ncbi.nlm.nih.gov . Проверено 5 января 2024 г.
- ^ Перейти обратно: а б с д Сарнат, Харви Б.; Марин-Гарсия, Хосе (май 2005 г.). «Патология митохондриальных энцефаломиопатий» . Канадский журнал неврологических наук . 32 (2): 152–166. дои : 10.1017/S0317167100003929 . ISSN 0317-1671 . ПМИД 16018150 . S2CID 1922603 .
- ^ Перейти обратно: а б Тобон А (декабрь 2013 г.). «Метаболические миопатии» . Континуум . 19 (6 мышечных заболеваний): 1571–1597. дои : 10.1212/01.CON.0000440660.41675.06 . ПМЦ 10563931 . ПМИД 24305448 . S2CID 11050341 .
- ^ Перейти обратно: а б Нури ЖБ, Заньоли Ф, Пети Ф, Маркорель П, Ранну Ф (май 2020 г.). «Нарушение работоспособности при метаболических миопатиях» . Научные отчеты . 10 (1): 8765. Бибкод : 2020NatSR..10.8765N . дои : 10.1038/s41598-020-65770-y . ПМК 7260200 . ПМИД 32472082 .
- ^ «Метаболизм питательных веществ у человека | Изучайте науку в Scitable» . www.nature.com . Проверено 5 апреля 2024 г.
- ^ «#540000 — МИТОХОНДРИАЛЬНАЯ МИОПАТИЯ, ЭНЦЕФАЛОПАТИЯ, МОЛОЧНЫЙ АЦИДОЗ И Инсультоподобные ЭПИЗОДЫ; МЕЛАС» . www.omim.org . Проверено 24 ноября 2023 г.
- ^ «#545000 — МИОКЛОНИЧЕСКАЯ ЭПИЛЕПСИЯ, СВЯЗАННАЯ С рваными КРАСНЫМИ ВОЛОКОНАМИ; MERRF» . www.omim.org . Проверено 24 ноября 2023 г.
- ^ «#530000 — СИНДРОМ КИРНС-СЕЙРА; KSS» . www.omim.org . Проверено 24 ноября 2023 г.
- ^ «Фенотипическая серия – PS157640 – CPEO – OMIM» . www.omim.org . Проверено 24 ноября 2023 г.
- ^ Перейти обратно: а б «Фенотипическая серия – PS603041 – Синдром истощения митохондриальной ДНК – OMIM» . www.omim.org . Проверено 24 ноября 2023 г.
- ^ «#500009 - МИТОХОНДРИАЛЬНАЯ МИОПАТИЯ, ДЕТСКАЯ, ТРАНЗИЕНТНАЯ; MMIT» . www.omim.org . Проверено 24 ноября 2023 г.
- ^ «# 551000 - МИТОХОНДРИАЛЬНАЯ МИОПАТИЯ, СМЕРТАЛЬНАЯ, ДЕТСКАЯ; LIMM» . www.omim.org . Проверено 30 ноября 2023 г.
- ^ Перейти обратно: а б с Легати, Андреа; Рейес, Аурелио; Чеккателли Берти, Камилла; Стелинг, Оливер; Марше, Сильвия; Ламперти, Костанца; Феррари, Альберто; Робинсон, Алан Дж.; Мюленхофф, Ульрих; Лилль, Роланд; Зевиани, Массимо; Гоффрини, Паола; Гецци, Даниэле (декабрь 2017 г.). «Новая доминантная мутация de novo в ISCU, связанная с митохондриальной миопатией» . Журнал медицинской генетики . 54 (12): 815–824. doi : 10.1136/jmedgenet-2017-104822 . ISSN 1468-6244 . ПМК 5740555 . ПМИД 29079705 .
- ^ "#255125 - МИОПАТИЯ С МОЛОЧНЫМ АЦИДОЗОМ, НАСЛЕДСТВЕННАЯ; ГМЛ" . www.omim.org . Проверено 24 ноября 2023 г.
- ^ «#500002 - МИТОХОНДРИАЛЬНАЯ МИОПАТИЯ ПРИ ДИАБЕТЕ» . www.omim.org . Проверено 24 ноября 2023 г.
- ^ «ДИАБЕТ И ГЛУХОТА, НАСЛЕДОВАННАЯ ПО МАТЕРИНСКОЙ СИСТЕМЕ; МИДД-ОМИМ» . www.omim.org . Проверено 2 марта 2024 г.
- ^ «#613076 — МИОПАТИЯ, МИТОХОНДРИАЛЬНАЯ ПРОГРЕССИВНАЯ, С ВРОЖДЕННОЙ КАТАРАКТОЙ И ЗАДЕРЖКОЙ РАЗВИТИЯ; ММПМЗ» . www.omim.org . Проверено 24 ноября 2023 г.
- ^ «Фенотипическая серия – PS600462 – Миопатия, лактоацидоз и сидеробластная анемия (MLASA) – OMIM» . www.omim.org . Проверено 30 ноября 2023 г.
- ^ «Аллельные варианты – 516060 – MT-ATP6 – OMIM» . www.omim.org . Проверено 30 ноября 2023 г.
- ^ «616209 - МИОПАТИЯ ИЗОЛИРОВАННАЯ МИТОХОНДРИАЛЬНАЯ АУТОСОМНО-ДОМИНАНТНАЯ; ИММД» . www.omim.org . Проверено 30 ноября 2023 г.
- ^ «#617675 — МИОПАТИЯ, МИТОХОНДРИИ И АТАКСИЯ; ММЯТ» . www.omim.org . Проверено 30 ноября 2023 г.
- ^ «#251900 - МИТОХОНДРИАЛЬНАЯ МИОПАТИЯ, ЭПИЗОДИЧЕСКАЯ, С ОПТИЧЕСКОЙ АТРОФЕЙ ИЛИ БЕЗ ИЛИ ОБРАТИЕМАЯ ЛЕЙКОЭНЦЕФАЛОПАТИЯ; МЕОАЛ» . www.omim.org . Проверено 30 ноября 2023 г.
- ^ «#251950 - МИТОХОНДРИАЛЬНАЯ МИОПАТИЯ С МОЛОЧНЫМ АЦИДОЗОМ; MMLA» . www.omim.org . Проверено 30 ноября 2023 г.
- ^ «251945 - МИТОХОНДРИАЛЬНАЯ МИОПАТИЯ С НАРУШЕНИЕМ МИТОХОНдриАЛЬНО-БЕЛКОВОГО ТРАНСПОРТА» . www.omim.org . Проверено 30 ноября 2023 г.
- ^ "*590000 - ТРАНСФЕРНАЯ РНК, МИТОХОНДРИАЛЬНАЯ, АЛАНИНА; МТТА" . www.omim.org . Проверено 30 ноября 2023 г.
- ^ «*590015 - ТРАНСФЕРНАЯ РНК, МИТОХОНДРИАЛЬНАЯ, АСПАРТИНОВАЯ КИСЛОТА; МТ-ТД» . www.omim.org . Проверено 30 ноября 2023 г.
- ^ «Аллельные варианты - 590095 - ТРАНСФЕРНАЯ РНК, МИТОХОНДРИЯ, ТРИПТОФАН; MT-TW - OMIM» . www.omim.org . Проверено 30 ноября 2023 г.
- ^ «Фенотипическая серия – PS607426 – Дефицит коэнзима Q10 – OMIM» . omim.org . Проверено 30 ноября 2023 г.
- ^ «Фенотипическая серия – PS252010 – Дефицит митохондриального комплекса I, ядерный тип – OMIM» . www.omim.org . Проверено 30 ноября 2023 г.
- ^ «Фенотипическая серия – PS252011 – митохондриальный комплекс II – OMIM» . www.omim.org . Проверено 30 ноября 2023 г.
- ^ «Аллельные варианты - 516020 - цитохром b комплекса III - OMIM» . www.omim.org . Проверено 30 ноября 2023 г.
- ^ «Фенотипическая серия – PS124000 – Дефицит митохондриального комплекса III, ядерный тип – OMIM» . www.omim.org . Проверено 1 декабря 2023 г.
- ^ «Фенотипическая серия – PS220110 – Дефицит митохондриального комплекса IV – OMIM» . www.omim.org . Проверено 30 ноября 2023 г.
- ^ «Фенотипическая серия – PS604273 – Дефицит митохондриального комплекса V – OMIM» . www.omim.org . Проверено 1 декабря 2023 г.
- ^ Штрауб В., Мерфи А., Удд Б. (август 2018 г.). «229-й международный семинар ENMC: Мышечные дистрофии пояса конечностей - Номенклатура и реформированная классификация Наарден, Нидерланды, 17-19 марта 2017 г.» . Нервно-мышечные расстройства . 28 (8): 702–710. дои : 10.1016/j.nmd.2018.05.007 . ПМИД 30055862 . S2CID 51865029 .
- ^ Бишелья Л., Зокколелла С., Торрако А., Пьемонтезе М.Р., Делл'Аглио Р., Амати А. и др. (июнь 2010 г.). «Новый локус 3p23-p25 для аутосомно-доминантной мышечной дистрофии пояса конечностей, LGMD1H» . Европейский журнал генетики человека . 18 (6): 636–641. дои : 10.1038/ejhg.2009.235 . ПМЦ 2987336 . ПМИД 20068593 .
- ^ «613530 - МЫШЕЧНАЯ ДИСТРОФИЯ, ПОЯСЫ КОНЕЧНОСТЕЙ, ТИП 1H; ПКМД1H» . www.omim.org . Проверено 30 ноября 2023 г.
- ^ Санакер, Питер С.; Томпу, Марина; Хоган, Ванесса Э.; Привет, Лангпин! Цулис, Харалампос; Хшановска-Лайтаулерс, Зофия М.А.; Тейлор, Роберт В.; Биндофф, Лоуренс А. (01 июня 2010 г.). «Различия в процессинге РНК лежат в основе тканеспецифического фенотипа миопатии ISCU» . Биохимия и биофизика Acta (BBA) - Молекулярные основы болезней . 1802 (6): 539–544. дои : 10.1016/j.badis.2010.02.010 . ISSN 0925-4439 .
- ^ Шауль, Орит (01 октября 2017 г.). «Как интроны усиливают экспрессию генов» . Международный журнал биохимии и клеточной биологии . Сращивание. 91 : 145–155. дои : 10.1016/j.biocel.2017.06.016 . ISSN 1357-2725 .
- ^ «Что такое некодирующая ДНК?: MedlinePlus Genetics» . medlineplus.gov . Проверено 11 апреля 2024 г.
- ^ Мораес, Коннектикут; Шанске, С.; Тричлер, Х.Дж.; Април, младший; Андреетта, Ф.; Бонилья, Э.; Шон, Э.А.; ДиМауро, С. (март 1991 г.). «Истощение мтДНК с переменной экспрессией в тканях: новая генетическая аномалия при митохондриальных заболеваниях» . Американский журнал генетики человека . 48 (3): 492–501. ISSN 0002-9297 . ПМК 1682992 . ПМИД 1998336 .
- ^ «#609560 — СИНДРОМ истощения митохондриальной ДНК 2 (миопатический тип); MTDPS2» . www.omim.org . Проверено 6 декабря 2023 г.
- ^ Перейти обратно: а б «Рваные красные мышечные волокна (идентификатор концепции: C3275417) — MedGen — NCBI» . www.ncbi.nlm.nih.gov . Проверено 6 декабря 2023 г.
- ^ Перейти обратно: а б Рифай З., Велле С., Камп С., Торнтон, Калифорния (январь 1995 г.). «Рваные красные волокна при нормальном старении и воспалительной миопатии». Анналы неврологии . 37 (1): 24–29. дои : 10.1002/ana.410370107 . ПМИД 7818253 . S2CID 23909958 .
- ^ Гомес А.П., Прайс Н.Л., Линг А.Дж., Мослехи Дж.Дж., Монтгомери М.К., Раджман Л. и др. (декабрь 2013 г.). «Снижение НАД(+) вызывает псевдогипоксическое состояние, нарушающее ядерно-митохондриальную связь во время старения» . Клетка . 155 (7): 1624–1638. дои : 10.1016/j.cell.2013.11.037 . ПМК 4076149 . ПМИД 24360282 .
- ^ Ким Дж. М. (ноябрь 2017 г.). «Миопатия, лекарства и митохондрии» . Журнал корейской медицинской науки . 32 (11): 1732–1733. дои : 10.3346/jkms.2017.32.11.1732 . ПМК 5639049 . ПМИД 28960021 .
- ^ Коултер Д.Л. (1 января 2014 г.). «Дефицит карнитина» . В: Аминофф М.Дж., Дарофф Р.Б. (ред.). Энциклопедия неврологических наук (второе изд.). Оксфорд: Академическая пресса. стр. 597–599. дои : 10.1016/B978-0-12-385157-4.00079-8 . ISBN 978-0-12-385158-1 . Проверено 30 ноября 2023 г.
- ^ Ли Ц, Ян С, Фэн Л, Чжао Ю, Су Ю, Лю Х и др. (2021). «Глутаровая ацидемия, патогенез и диетотерапия» . Границы в питании . 8 : 704984. дои : 10.3389/fnut.2021.704984 . ПМЦ 8714794 . ПМИД 34977106 .
- ^ Бехин А., Аквавива-Бурден С., Суваннанорат С., Штрейхенбергер Н., Аттариан С., Бассес Г. и др. (март 2016 г.). «Множественный дефицит ацил-КоА-дегидрогеназы (MADD) как причина излечимых метаболических заболеваний с поздним началом». Ревю Неврологии . 172 (3): 231–241. дои : 10.1016/j.neurol.2015.11.008 . ПМИД 27038534 .
- ^ Энрикес Б.Дж., Катрин Джентофт Олсен Р., Гомес С.М., Бросс П. (апрель 2021 г.). «Флавопротеин-переносчик электронов и его роль в митохондриальном энергетическом обмене в норме и при заболеваниях» . Джин . 776 : 145407. doi : 10.1016/j.gene.2021.145407 . ПМЦ 7949704 . ПМИД 33450351 .
- ^ «#616839 — НЕПЕРЕНОСИМОСТЬ УПРАЖНЕНИЙ, РИБОФЛАВИН-ОТВЕТСТВЕННЫЙ; RREI» . www.omim.org . Проверено 24 ноября 2023 г.
- ^ Шозер, BGH; Мюллер-Хёккер, Дж.; Хорват, Р.; Гемпель, К.; Понгратц, Д.; Лохмюллер, Х.; Мюллер-Фельбер, В. (октябрь 2007 г.). «Болезнь накопления гликогена у взрослых, тип 2: пересмотр клинико-патологического фенотипа» . Невропатология и прикладная нейробиология . 33 (5): 544–559. дои : 10.1111/j.1365-2990.2007.00839.x . ISSN 0305-1846 . ПМИД 17573812 . S2CID 25822083 .
- ^ Перейти обратно: а б с Саха, Мадхурима; Редди, Хемакумар М.; Салих, Мустафа А.; Эстрелла, Элисия; Джонс, Майкл Д.; Мицухаси, Сатоми; Чо, Кён-А; Сузуки-Хатано, Сильвели; Риццо, Скайлар А.; Хамад, Муддатир Х.; Мухтар, Маовия М.; Хамед, Ахлам А.; Элсид, Маха А.; Лек, Монкол; Валканас, Элиза (01 ноября 2018 г.). «Влияние дефицита PYROXD1 на клеточное дыхание и корреляции с генетическим анализом мышечной дистрофии пояса конечностей в Саудовской Аравии и Судане» . Физиологическая геномика . 50 (11): 929–939. doi : 10.1152/физиологгеномика.00036.2018 . ISSN 1531-2267 . ПМК 6293114 . ПМИД 30345904 .
- ^ Вирапандиян А., Шаши В., Цзян Й.Х., Галлентайн В.Б., Шох К., Смит Э.К. (декабрь 2010 г.). «Псевдометаболическая картина дистрофинопатии вследствие миссенс-мутации» . Мышцы и нервы . 42 (6): 975–979. дои : 10.1002/mus.21823 . ПМК 5506871 . ПМИД 21104870 .
- ^ Бартон, Элизабет Р.; Пачак, Кристина А.; Стоппель, Уитни Л.; Канг, Питер Б. (29 июля 2020 г.). «Связующие узы: функциональные кластеры при мышечной дистрофии конечностей» . Скелетная мышца . 10 (1): 22. дои : 10.1186/s13395-020-00240-7 . ISSN 2044-5040 . ПМЦ 7389686 . ПМИД 32727611 .
- ^ Сайнио, Маркус Т.; Вялипакка, Салла; Ринальди, Бруно; Лапатто, Хелена; Паэтау, Андерс; Оянен, Симо; Брильанте, Вирджиния; Джокела, Ману; Хуовинен, Санна; Ауранен, Мари; Пальмио, Джоанна; Фриан, Сильви; Иликаллио, Эмиль; Удд, Бьярн; Тюнисмаа, Хна (февраль 2019 г.). «Рецессивные мутации PYROXD1 вызывают у взрослых мышечную дистрофию конечностей-поясного типа» . Журнал неврологии . 266 (2): 353–360. дои : 10.1007/s00415-018-9137-8 . ISSN 1432-1459 . ПМК 6373352 . ПМИД 30515627 .