Лице-лопаточно-плечевая мышечная дистрофия
| Лице-лопаточно-плечевая мышечная дистрофия | |
|---|---|
| Другие имена | Мышечная дистрофия Ландузи-Дежерина, ФСГМД, ФСГ |
 | |
| Диаграмма, показывающая мышцы, обычно поражаемые при ЛЛПД. | |
| Произношение | |
| Специальность | Неврология , нервно-мышечная медицина |
| Симптомы | Слабость лица, крыло лопатки, опущение стопы. |
| Осложнения | Хроническая боль, сухость глаз и нестабильность плеч; реже заболевания сетчатки, сколиоз и дыхательная недостаточность. |
| Обычное начало | Возраст 15 – 30 лет |
| Продолжительность | Пожизненный |
| Типы | Обычно классифицируется по генетической причине (ЛЛПД1, ЛЛПД2). Иногда классифицируется по проявлению заболевания (например, начало в младенческом возрасте) |
| Причины | Генетический (наследственная или новая мутация) |
| Факторы риска | Мужской пол, степень генетической мутации |
| Метод диагностики | Генетическое тестирование |
| Дифференциальный диагноз | Поясно- конечностная мышечная дистрофия (особенно кальпаинопатия ), болезнь Помпе , митохондриальная миопатия , полимиозит [2] |
| Управление | Физиотерапия, корсет, реконструктивная хирургия |
| Медикамент | Клинические испытания продолжаются |
| Прогноз | Прогрессирующая, неизмененная продолжительность жизни |
| Частота | До 1/8,333 [2] |
Лице-лопаточно-плечевая мышечная дистрофия ( ЛЛПД ) — разновидность мышечной дистрофии , группа наследственных заболеваний , вызывающих дегенерацию мышц и прогрессирующую слабость . Судя по названию , ЛЛПД имеет тенденцию последовательно ослаблять мышцы лица , которые позиционируют лопатку те , , и те, которые лежат над плечевой костью плеча. [2] [3] Эти области могут быть сохранены, но обычно поражаются мышцы других областей, особенно груди , живота , позвоночника и голени . практически любая скелетная мышца На поздних стадиях заболевания может быть поражена . Часто встречаются аномально расположенные лопатки, называемые «крылатыми» , а также неспособность поднять стопу, известная как « опущение стопы ». Обе стороны тела часто поражаются неодинаково. Слабость обычно проявляется в возрасте 15–30 лет. [4] ЛЛПД также может вызывать потерю слуха и аномалии кровеносных сосудов в задней части глаза .
ЛЛПД вызван генетической мутацией , приводящей к нарушению регуляции DUX4 гена . [5] В норме DUX4 экспрессируется (т.е. включается ) только в избранных тканях человека, особенно в очень молодых эмбрионах . В остальных тканях он репрессирован (т. е. выключен). [6] [7] При ЛЛПД эта репрессия в мышечной ткани нарушается, что обеспечивает спорадическую экспрессию DUX4 на протяжении всей жизни. Делеция ДНК является в области, окружающей DUX4, причинной мутацией в 95% случаев, называемой « сокращением D4Z4 » и определяющей тип 1 ЛЛПД (ЛЛПД1). [8] ЛЛПД, вызванный другими мутациями, относится к ЛЛПД 2 типа (ЛЛПД2). Для развития заболевания также необходим аллель 4qA , который является распространенной вариацией ДНК рядом с DUX4 . Шансы на передачу ребенку сокращения D4Z4 с аллелем 4qA составляют 50% ( аутосомно-доминантный тип ); [2] в 30% случаев мутация возникла спонтанно. [4] Мутации FSHD вызывают неадекватную DUX4 репрессию , распаковывая ДНК вокруг DUX4 , делая ее доступной для копирования в информационную РНК (мРНК). Аллель 4qA стабилизирует мРНК DUX4 , что позволяет использовать ее для производства DUX4 белка . [9] Белок DUX4 является модулятором сотен других генов, многие из которых участвуют в работе мышц. [2] [5] Как эта генетическая модуляция вызывает повреждение мышц, остается неясным. [2]
Признаки, симптомы и диагностические тесты могут указывать на ЛЛПД; генетическое тестирование обычно обеспечивает окончательный диагноз. [2] ЛЛПД может быть предположительно диагностирован у человека с признаками/симптомами и установленным семейным анамнезом. Ни одно вмешательство не доказало свою эффективность в замедлении прогрессирования слабости. [10] Скрининг позволяет на ранней стадии выявить и принять меры при различных осложнениях заболевания. Симптомы можно устранить с помощью физиотерапии, корсета и реконструктивной хирургии, такой как хирургическая фиксация лопатки к грудной клетке. [11] ЛЛПД поражает до 1 из 8333 человек, [2] помещая ее в тройку наиболее распространенных мышечных дистрофий с миотонической дистрофией и мышечной дистрофией Дюшенна . [12] [13] Прогноз варьируется. Многие существенно не ограничены в повседневной деятельности, тогда как инвалидная коляска или самокат требуются в 20% случаев. [14] Продолжительность жизни не изменяется, хотя смерть редко может быть связана с дыхательной недостаточностью, вызванной ЛЛПД. [15]
Впервые ЛЛПД был выделен как заболевание в 1870-х и 1880-х годах, когда французские врачи Луи Теофиль Жозеф Ландузи и Жозеф Жюль Дежерин наблюдали за семьей, затронутой этим заболеванием, отсюда и первоначальное название « мышечная дистрофия Ландузи-Дежерина» . Их работе предшествовали описания вероятных отдельных случаев ЛЛПД. [16] [17] [18] Значение сокращения D4Z4 на хромосоме 4 было установлено в 1990-х годах. Ген DUX4 был открыт в 1999 году, его экспрессия и токсичность были обнаружены в 2007 году, а в 2010 году был выяснен генетический механизм, вызывающий его экспрессию. В 2012 году был идентифицирован ген, наиболее часто мутирующий при ЛЛПД2. В 2019 году первый препарат, предназначенный для противодействия экспрессии DUX4, вступил в клинические испытания. [19]
Признаки и симптомы
[ редактировать ]Классически слабость развивается в лице, затем в плечевом поясе , затем в плече . [10] Эти мышцы можно сохранить, но обычно поражаются другие мышцы. Порядок включения мышц может стать причиной появления слабости, «нисходящей» от лица к ногам. [10] Распространение и степень мышечной слабости чрезвычайно вариабельны, даже у однояйцевых близнецов. [20] [21] Скелетно-мышечная боль встречается очень часто, чаще всего описывается в шее, плечах, пояснице и задней части колена. [22] [4] Усталость также является распространенным явлением. [4] Мышечная слабость обычно становится заметной на одной стороне тела раньше, чем на другой, что является отличительным признаком заболевания. [14] Мышцы правого плеча и руки поражаются чаще, чем мышцы левой верхней конечности, независимо от руки . [23] : 139 [24] [25] [26] В противном случае ни одна из сторон тела не подвергалась большему риску. Обычно симптомы появляются в возрасте 15–30 лет, хотя может возникнуть и во взрослом возрасте. [4] Инфантильное начало (также называемое ранним началом), определяемое как начало заболевания в возрасте до 10 лет, встречается у 10% заболевших людей. [10] FSHD1 с очень большой делецией D4Z4 ( EcoRI 10-11 kb ) более тесно связан с инфантильным началом и тяжелой слабостью. [27] Отсутствие или почти полное отсутствие симптомов не является чем-то необычным и достигает до 30% лиц, несущих мутации, в некоторых семьях с ЛЛПД1. [10] В среднем ЛЛПД2 проявляется на 10 лет позже, чем ЛЛПД1. [28] В противном случае FSHD1 и FSHD2 неотличимы по слабости. [27] Прогрессирование заболевания происходит медленно, и нередки длительные статические фазы, в которых прогрессирование не наблюдается. [29] Реже отдельные мышцы быстро изнашиваются в течение нескольких месяцев. [2] Бремя симптомов ЛЛПД обычно более серьезное, чем кажется людям, не имеющим этого заболевания. [30] [31] [32] [33]
Лицо
[ редактировать ]Слабость мышц лица является наиболее отличительным признаком ЛЛПД. [4] Обычно это самый ранний признак, хотя редко это первая жалоба. [4] По крайней мере, легкая слабость лицевых мышц может быть обнаружена у 90% и более пациентов с ЛЛПД. [29] [24] Одним из наиболее распространенных недостатков является неспособность закрыть веки, что может привести к сну с открытыми веками и сухости глаз. [4] Задействованной мышцей является круговая мышца глаза . [4] Еще одним распространенным недостатком является неспособность сжать губы, что приводит к невозможности сморщить, свистнуть или надуть воздушный шарик. [4] Задействованной мышцей является круговая мышца рта . [4] Третий распространенный недостаток — неспособность поднять уголки рта, вызывая «горизонтальную улыбку», которая больше похожа на ухмылку. [4] За это отвечает большая скуловая мышца. [4]
Слабость лицевых мышц способствует затруднению произношения слов . [34] Выражение лица может выглядеть унылым, высокомерным, сварливым или утомленным. [4] Мышцы, отвечающие за жевание и движение глаз, не поражаются. [24] [14] Затруднение глотания нетипично, хотя может возникнуть в запущенных случаях, что, по крайней мере, частично связано со слабостью лицевых мышц. [35] [34] ЛЛПД обычно прогрессирует, но не установлено, является ли слабость лицевых мышц прогрессирующей или стабильной на протяжении всей жизни. [36]
Плечо, грудь и рука
[ редактировать ]
После лицевой слабости обычно развивается слабость мышц груди и мышц, простирающихся от лопатки до грудной клетки. Симптомы, связанные с плечом, такие как трудности при работе с руками над головой, являются первоначальными жалобами в 80% случаев. [24] [14] Преимущественно передние зубчатые мышцы , а также средняя и нижняя мышцы трапеций ; поражаются [4] верхняя часть трапеции часто сохраняется. [14] Слабость трапециевидных мышц приводит к тому, что лопатки поворачиваются вниз и вытягиваются , что приводит к появлению крылатых лопаток , горизонтальным ключицам и покатым плечам; отведение руки нарушено. Слабость передних зубчатых мышц ухудшает сгибание рук , а ухудшение крыльев может наблюдаться при надавливании на стену. Мышцы, простирающиеся от лопатки до руки, обычно сохраняются, включая дельтовидные мышцы и мышцы вращательной манжеты плеча . [37] [38] Позже может поражаться дельтовидная мышца, особенно верхняя часть. [4]
Сильная атрофия мышц может привести к тому, что кости и сохранившиеся мышцы плеч станут очень заметными, характерным примером является симптом «многохолма», возникающий при поднятии руки. [4] Первый «бугор» или шишка — это верхний угол лопатки, который выглядит «грыжей» вверх и над грудной клеткой. Второй холм — это сустав переменного тока , расположенный между атрофированной верхней частью трапеции и атрофированной верхней дельтовидной мышцей. Третий холм — это нижняя дельтовидная мышца, отличающаяся от атрофированной верхней дельтовидной мышцы и атрофированной плечевой мышцы. [4] Слабость и боль в плече, в свою очередь, могут привести к нестабильности плеча, например, к рецидивирующему вывиху , подвывиху или перемещению головки плечевой кости вниз . [39]
Также поражается грудная клетка, особенно части большой грудной мышцы , которые соединяются с грудиной и ребрами. Реже поражается часть, которая соединяется с ключицей. Такая картина атрофии мышц может способствовать появлению заметной горизонтальной передней подмышечной складки . [40] [4] За пределами этой точки заболевание не прогрессирует дальше в 30% семейных случаев. [24] [14] После слабости верхней части туловища слабость может «спуститься» на плечи ( двуглавую мышцу и особенно трехглавую мышцу ). [24] Предплечья обычно сохраняются, в результате чего внешний вид некоторых сравнивают с вымышленным персонажем Попаем . [4] хотя при поражении предплечий на поздних стадиях заболевания разгибатели запястья . чаще поражаются [24]
Нижняя часть тела и туловище
[ редактировать ]После верхней части тела слабость может появиться либо в тазу, либо она «пропускает» таз и затрагивает переднюю большеберцовую мышцу (мышцу голени), вызывая опущение стопы . Один автор считает, что мышцы таза и бедра поражаются последней группой. [24] Слабость таза может проявляться симптомом Тренделенбурга . [4] Слабость задней части бедра (подколенных сухожилий) встречается чаще, чем слабость передней части бедра (квадрицепса). [4] В более тяжелых случаях, особенно у детей с ЛЛПД, может наблюдаться наклон таза вперед и связанное с ним переразгибание коленей. [41]
Слабость может также возникать в мышцах живота и параспинальных мышцах , что может проявляться в виде выпирающего живота и поясничного гиперлордоза . [2] [4] Слабость в животе может привести к неспособности приседать или неспособности повернуться с одной стороны на другую, лежа на спине. [4] Из прямых мышц живота преимущественно поражается нижняя часть, что проявляется положительным симптомом Бивора . [4] [2] В запущенных случаях слабость разгибателей шеи может привести к наклону головы к груди, что называется опущением головы. [24]
Немышечный
[ редактировать ]
Наиболее частым некостно-мышечным проявлением ЛЛПД являются аномалии мелких артерий ( артериол ) сетчатки . Извитость артериол наблюдается примерно у 50% больных ЛЛПД. Менее распространенные аномалии артериол включают телеангиэктазии и микроаневризмы . [42] [43] Эти аномалии артериол обычно не влияют на зрение или здоровье, хотя их тяжелая форма имитирует болезнь Коата , состояние, которое встречается примерно в 1% случаев ЛЛПД и чаще связано с большими делециями 4q35. [2] [44] Высокочастотная нейросенсорная тугоухость может возникнуть у людей с большой делецией 4q35, но в остальном она не более распространена по сравнению с общей популяцией. [2] Большая делеция 4q35 может привести к другим редким проявлениям. [45]
Может возникнуть сколиоз , который, как полагают, является результатом слабости мышц живота, разгибателей бедра и позвоночника. [46] [47] И наоборот, сколиоз можно рассматривать как компенсаторный механизм слабости. [46] Дыхание может быть нарушено, что связано с кифосколиозом и использованием инвалидной коляски; оно наблюдается у трети пациентов, пользующихся инвалидными колясками. [2] Однако аппаратная поддержка (ночная или дневная) необходима только в 1% случаев. [2] [48] Хотя есть сообщения о повышенном риске сердечных аритмий, общее мнение заключается в том, что сердце не затрагивается. [14]
Генетика
[ редактировать ]
Генетика ЛЛПД сложна. [2] ЛЛПД и миотонические дистрофии имеют уникальные генетические механизмы, существенно отличающиеся от остальных генетических миопатий. [49] Ген DUX4 является центром генетики ЛЛПД. В норме полноразмерный белок DUX4 (DUX4-fl) экспрессируется во время раннего эмбриогенеза , в ткани яичка взрослых и в тимусе ; во всех других тканях он подавляется . [50] При ЛЛПД в мышечной ткани происходит нарушение репрессии DUX4 и продолжается выработка белка DUX4-fl, который токсичен для мышц. [2] [8] [50] Механизм неудавшейся DUX4 репрессии гипометилировании DUX4 заключается в и окружающей его ДНК на кончике хромосомы 4 (4q35), что обеспечивает в информационную транскрипцию DUX4 РНК ( мРНК). Некоторые мутации могут привести к заболеванию, в результате чего ЛЛПД подразделяется на ЛЛПД 1-го типа (ЛЛПД1) и ЛЛПД 2-го типа (ЛЛПД2). [27] Заболевание может возникнуть только тогда, когда мутация присутствует в сочетании с избранными, часто встречающимися вариациями 4q35, называемыми гаплотипов полиморфизмами , которые условно можно разделить на группы 4qA и 4qB. [51] Полиморфизм гаплотипа 4qA, часто называемый аллелем 4qA , необходим для заболевания, поскольку он содержит последовательность полиаденилирования , которая позволяет мРНК DUX4 противостоять деградации достаточно долго для трансляции в белок DUX4. [8]
DUX4 и массив повторов D4Z4
[ редактировать ]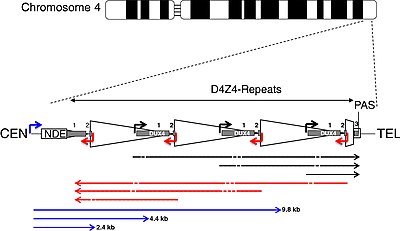
| CEN | центромерный конец | ТЕЛ. | теломерный конец |
| коробка неразрушающего контроля | неудаленный элемент | НЕТ | полиаденилирования сайт |
| треугольник | D4Z4 повтор | трапеция | частичный повтор D4Z4 |
| белая коробка | ПЛАМ | серые коробки | DUX4 экзоны 1, 2, 3 |
| стрелки | |||
| угол | промоутеры | прямой | Транскрипты РНК |
| черный | смысл | красный | антисмысловой |
| синий | ДБЭ-Т | тире | сайты для игры в кости |
DUX4 находится в массиве макросателлитных повторов D4Z4, который представляет собой последовательность ДНК, состоящую из переменного числа тандемно повторяющихся больших сегментов ДНК (т.е. повторов). Массив повторов D4Z4 расположен на кончике большого плеча хромосомы 4 , сокращенно называемого «4q35». [52] Каждый повтор D4Z4 имеет длину 3,3 тысячи пар оснований (т.п.н.) и является местом эпигенетической регуляции, содержащим как гетерохроматиновые , так и эухроматиновые структуры. [53] [54] При ЛЛПД структура гетерохроматина теряется, превращаясь в эухроматин. [53] который состоит из меньшего метилирования ДНК и измененного метилирования гистонов . [55] Паттерны метилирования гистонов немного различаются при ЛЛПД1 и ЛЛПД2. [55]
Субтеломерная область хромосомы 10q содержит структуру тандемных повторов, высоко гомологичную (идентичную на 99%) 4q35. [8] [51] содержащие «D4Z4-подобные» повторы с областями, кодирующими белок, идентичными DUX4 (повторы D10Z10 и DUX4L10 соответственно). [8] [56] Поскольку в 10q обычно отсутствует последовательность полиаденилирования, он обычно не участвует в заболевании. Однако хромосомные перестройки могут происходить между массивами повторов 4q и 10q, и участие в заболевании возможно, если повтор 4q D4Z4 и сигнал полиаденилирования передаются на 10q. [57] [8] [58] или если перестановка вызывает FSHD1.
Типы массивов повторов D4Z4 подразделяются на аллели 4qA и 4qB, причем только аллели 4qA вызывают заболевание. Аллели 4qA определяются специфической последовательностью ДНК, расположенной непосредственно ниже массива повторов D4Z4: областью из 260 пар оснований, называемой pLAM, за которой следует бета-сателлитная область из 6200 пар оснований. [9] [59] Аллели 4qA и 4qB вместе можно разделить как минимум на 17 типов. [51] на основе ДНК выше массива повторов D4Z4, наличия/отсутствия сайтов рестрикции в D4Z4, размера последнего элемента повтора D4Z4 и ДНК, присутствующей ниже массива повторов D4Z4. [59] Например, наиболее распространенный аллель 4qA, 4A161, имеет 161 нуклеотид в области выше массива повторов D4Z4. [27] и, в свою очередь, подразделяются на 4A161S и 4A161L (короткие и длинные), которые характеризуются фланкирующими повторяющимися единицами D4Z4 длиной 300 нуклеотидов и 1900 нуклеотидов соответственно. [59]
DUX4 состоит из трех экзонов . Экзоны 1 и 2 находятся в каждом повторе; только экзон 1 кодирует белок DUX4. Экзон 3 находится в теломерной области pLAM до последнего частичного повтора, [8] [7] и его длина может варьироваться в зависимости от типа аллеля 4qA. [60] Сигнал полиаденилирования находится внутри экзона 3. Поскольку экзон 3 и содержащий его сигнал полиаденилирования не содержатся в каждом повторе D4Z4, только последний повтор D4Z4 массива повторов D4Z4 может кодировать стабильный транскрипт мРНК для продукции белка DUX4. [50] Эти транскрипты можно сплайсировать несколькими различными способами с образованием зрелой РНК. Один из этих транскриптов кодирует только часть белка DUX4, называемую DUX4-s (DUX4-short). [50] DUX4-s обнаруживается в небольших количествах во многих здоровых тканях, включая здоровые мышцы; его функция не совсем ясна. [50] Остальные версии транскрипта кодируют полноразмерный белок DUX4 (DUX4-fl), отличаясь только некодирующими участками. [50] Только определенные версии этих зрелых РНК, кодирующих DUX4-fl, вовлечены в FSHD. [50] Помимо области DUX4, из массива повторов D4Z4 производятся множественные транскрипты РНК, как смысловые, так и антисмысловые, некоторые из которых могут подвергаться деградации в определенных участках с образованием si-подобных малых РНК. [27] Некоторые транскрипты, которые происходят центромерно от массива повторов D4Z4 на неудаленном элементе (NDE), называемые транскриптами регуляторного элемента D4Z4 (DBE-T), могут играть роль в дерепрессии DUX4 . [27] [61] Один из предполагаемых механизмов заключается в том, что DBE-T приводит к рекрутированию группы триторакса белка Ash1L , увеличению метилирования H3K36me2 и, в конечном итоге, дерепрессии генов 4q35. [62]
Метилирование ДНК | ||||
|---|---|---|---|---|
| ФСД1 | ↓ | ↑ | ↓ | |
| ФШД2* | ↓ | ↑ | ↓ | ↑ |
| *Профили хроматина не полностью характеризуется DNMT3B мутацией [55] | ||||
ФСД1
[ редактировать ]ЛЛПД, включающий делецию повторов D4Z4 (так называемое «сокращение D4Z4») в 4q, классифицируется как ЛЛПД1, на который приходится 95% случаев ЛЛПД. [2] Обычно хромосома 4 включает от 11 до 150 повторов D4Z4. [53] [8] В FSHD1 имеется от 1 до 10 повторов D4Z4. [8] Количество повторов примерно обратно пропорционально тяжести заболевания. А именно, у пациентов с 8–10 повторами, как правило, самые легкие проявления, иногда без симптомов; у пациентов с 4–7 повторами заболевание средней степени тяжести и сильно варьирует; а у пациентов с 1–3 повторами чаще наблюдается тяжелое, атипичное заболевание с ранним началом. [63] Удаление всего массива повторов D4Z4 не приводит к FSHD, поскольку тогда не остается полных копий DUX4, которые могут быть экспрессированы, хотя возникают и другие врожденные дефекты. [64] [8] Одного сокращенного массива повторов D4Z4 с прилегающим аллелем 4qA достаточно, чтобы вызвать заболевание, поэтому наследование является аутосомно-доминантным . Мутации de novo (новые) возникают в 10–30% случаев. [4] до 40% из них демонстрируют соматический мозаицизм . [14] У человека с мозаичной ЛЛПД тяжесть заболевания коррелирует с долей клеток, несущих мутацию. [14]
Было высказано предположение, что FSHD1 подвергается ожиданию - феномену, в первую очередь связанному с нарушениями тринуклеотидных повторов , при которых проявления заболевания ухудшаются с каждым последующим поколением. [65] По состоянию на 2019 год необходимы более детальные исследования, чтобы окончательно показать, возникает ли ожидание или нет. [66] Если ожидание действительно возникает при ЛЛПД, механизм отличается от механизма нарушений тринуклеотидных повторов, поскольку повторы D4Z4 намного крупнее тринуклеотидных повторов, недостаток повторов (а не избыток) вызывает заболевание, а размер массива повторов при ЛЛПД стабилен на протяжении всего периода. поколения. [67]
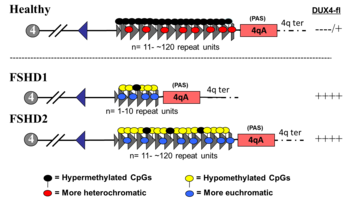
ФШД2
[ редактировать ]ЛЛПД с размером повтора массива D4Z4 11 или более классифицируется как ЛЛПД2, что составляет 5% случаев ЛЛПД. [2] Аллель 4qA по-прежнему необходим, а соседний с ним массив повторов D4Z4 обычно укорочен и содержит менее 30 повторов. Для FSHD2 необходима деактивирующая мутация одного из нескольких генов метилирования ДНК, что способствует гипометилированию пограничного укороченного массива повторов D4Z4, при котором генетический механизм сходится с FSHD1. [68] [10] По крайней мере, 85% случаев ЛЛПД2 связаны с мутациями в гене SMCHD1 (структурное сохранение гибкого шарнирного домена хромосомы, содержащего 1) на хромосоме 18 . [10] Специфические мутации SMCHD1 также связаны с босмаринией и синдромом микрофтальмии . [55] Другой причиной ЛЛПД2 является мутация DNMT3B (ДНК-метилтрансфераза 3B). [69] [70] Мутации в DNMT3B также могут вызывать синдром ИКФ . [55] По состоянию на 2020 год первые данные указывают на то, что третьей причиной ЛЛПД2 является мутация гена LRIF1 , который кодирует белок -лиганд-зависимый фактор взаимодействия с ядерными рецепторами 1 (LRIF1). [71] Известно, что LRIF1 взаимодействует с белком SMCHD1. [71] По состоянию на 2019 год предполагается, что мутация дополнительных неопознанных генов может вызвать ЛЛПД2. [2]
Для FSHD2, связанного с мутацией SMCHD1 или DNMT3B , только один аллель должен быть аномальным. Эти гены, вызывающие FSHD2, не расположены рядом с необходимым массивом D4Z4/аллелем 4qA в геноме и, таким образом, наследуются независимо , что приводит к двуаллельному дигенному типу наследования. Например, один родитель без FSHD может передать мутацию SMCHD1 , а другой родитель, также без FSHD, может передать укороченный по границе аллель D4Z4/4qA, вынашивая ребенка с FSHD2. [68] [70] Для FSHD2, связанного с мутацией LRIF1 , LRIF1 , что теоретически приводит к еще более сложному типу наследования, называемому триаллейным дигенным. необходимо мутировать оба аллеля [71] [72]
Два конца спектра заболеваний
[ редактировать ]ЛЛПД1 и ЛЛПД2 традиционно рассматривались как отдельные объекты с разными генетическими причинами (хотя нижестоящие генетические механизмы сливаются). [73] Альтернативно, генетические причины ЛЛПД1 и ЛЛПД2 можно рассматривать как факторы риска , каждый из которых вносит свой вклад в спектр заболеваний ЛЛПД. [73] Нередко кажется, что пострадавший человек имеет вклад и от того, и от другого. [63] Например, у людей с ЛЛПД2, хотя у них нет аллеля 4qA с числом повторов D4Z4 менее 11, у них все же есть один из них меньше 30 (меньше верхнего предела, наблюдаемого в общей популяции), что позволяет предположить, что большое количество повторов повторов D4Z4 может предотвратить последствия мутации SMCHD1 . [63] [10] Могут потребоваться дальнейшие исследования для определения верхнего предела повторов D4Z4 при ЛЛПД2. [63]
У пациентов с ЛЛПД1 и ЛЛПД2, то есть имеющих 10 или менее повторов с соседним аллелем 4qA и мутацией SMCHD1 , заболевание проявляется более тяжело, демонстрируя, что эффекты каждой мутации аддитивны. [74] Комбинированное представление FSHD1/FSHD2 чаще всего встречается у пациентов с 9–10 повторами. Возможное объяснение состоит в том, что значительная часть населения в целом имеет 9–10 повторов, при которых заболевание трудно обнаружить или вообще отсутствует, однако при аддитивном эффекте мутации SMCHD1 симптомы достаточно серьезны для постановки диагноза. [63] Размер повторов 9–10 можно рассматривать как зону перекрытия между FSHD1 и FSDH2. [63]
Патофизиология
[ редактировать ]Молекулярный
[ редактировать ]
По состоянию на 2020 год, похоже, существует консенсус в отношении того, что аберрантная экспрессия DUX4 в мышцах является причиной ЛЛПД. [75] DUX4 экспрессируется в чрезвычайно небольших количествах, обнаруживаемых в 1 из каждых 1000 незрелых мышечных клеток (миобластов) , количество которых, по-видимому, увеличивается после созревания миобластов, отчасти потому, что клетки сливаются по мере их созревания, и единственное ядро, экспрессирующее DUX4, может обеспечить DUX4. белок к соседним ядрам из слившихся клеток. [76]
Остается область активных исследований того, как белок DUX4 вызывает повреждение мышц. [77] Белок DUX4 является фактором транскрипции, который регулирует многие другие гены. Некоторые из этих генов участвуют в апоптозе , например, p53 , p21 , MYC и β-катенин , и действительно кажется, что белок DUX4 делает мышечные клетки более склонными к апоптозу. Другие гены, регулируемые белком DUX4, участвуют в окислительном стрессе , и действительно кажется, что экспрессия DUX4 снижает толерантность мышечных клеток к окислительному стрессу. Различия в способности отдельных мышц справляться с окислительным стрессом могут частично объяснить характер поражения мышц при ЛЛПД. Белок DUX4 подавляет многие гены, участвующие в развитии мышц, включая MyoD , миогенин , десмин и PAX7 , и действительно, экспрессия DUX4 , как было показано, снижает мышечных клеток пролиферацию, дифференцировку и слияние . Белок DUX4 регулирует несколько генов, которые участвуют в контроле качества РНК , и действительно DUX4 вызывает накопление РНК с последующим апоптозом. было показано, что экспрессия [75] Эстроген , по-видимому, играет роль в изменении воздействия белка DUX4 на дифференцировку мышц, что может объяснить, почему женщины страдают от этого в меньшей степени, чем мужчины, хотя это остается недоказанным. [78]
клеточная реакция на гипоксию В одном исследовании сообщалось, что является основной причиной гибели мышечных клеток, вызванной белком DUX4. Факторы , индуцируемые гипоксией (HIF), активируются белком DUX4, что, возможно, вызывает патологическую передачу сигналов, приводящую к гибели клеток. [79]
Другое исследование показало, что экспрессия DUX4 в мышечных клетках приводит к рекрутированию и изменению фиброзных / жировых клеток-предшественников, что помогает объяснить, почему мышцы заменяются жировой и фиброзной тканью . [76]
Единственное исследование показало участие RIPK3 в гибели клеток, опосредованной DUX4. [80]
Гистология мышц
[ редактировать ]
В отличие от других мышечных дистрофий, ранние биопсии мышц показывают лишь легкую степень фиброза , гипертрофию мышечных волокон и смещение ядер от периферии миоволокон (центральное зарождение). [27] Чаще встречается воспаление. [27] Может наблюдаться эндомизиальное воспаление, состоящее в основном из CD8+ Т-клеток , хотя эти клетки, по-видимому, не вызывают непосредственно гибели мышечных волокон. [27] Эндомизиальные кровеносные сосуды могут быть окружены воспалением, которое относительно уникально для ЛЛПД, и это воспаление содержит CD4+ Т-клетки . [27] Воспаление сменяется отложением жира (жировой инфильтрацией), затем фиброзом. [81] [27] Отдельные мышечные волокна могут выглядеть извитыми, изъеденными молью и, особенно, дольчатыми. [82]
Схема вовлечения мышц
[ редактировать ]Почему при ЛЛПД преимущественно поражаются определенные мышцы, остается неизвестным. При ЛЛПД наблюдается множество тенденций вовлечения, возможно, намекающих на лежащую в основе патофизиологию. Отдельные мышцы могут ослабевать, в то время как соседние мышцы остаются сильными. [83] Мышцы правого плеча и руки поражаются чаще, чем мышцы левых верхних конечностей, такая же картина наблюдается при синдроме Поланда и наследственной невралгической амиотрофии ; это может отражать генетический, онтогенетический/анатомический или функциональный механизм. [24] [25] Дельтовидная мышца часто сохраняется, чего не наблюдается ни при каких других состояниях, поражающих мышцы вокруг лопатки. [36]

Медицинская визуализация (КТ и МРТ) показала поражение мышц, которое иначе не было бы очевидно. [37]
- Одно МРТ-исследование показывает, большая круглая мышца . что чаще всего поражается [38]
- , полуперепончатая мышца входящая в состав подколенных сухожилий . Обычно поражается [25] [84] [85] по мнению одного автора, это «наиболее часто и серьезно поражаемая мышца». [2]
- Из четырехглавых мышц прямая мышца бедра . преимущественно поражается [84]
- Из икроножных мышц преимущественно поражается медиальный отдел; [84] [85]
- мышца Подвздошно-поясничная , мышца-сгибатель бедра, очень часто сохраняется. [85] [2]
Ретинопатия
[ редактировать ]При ЛЛПД часто обнаруживаются извилистость артериол сетчатки, реже микроаневризмы и телеангиэктазии. [42] Аномалий со стороны капилляров и венул не наблюдается. [42] Одна из теорий, почему артериолы поражаются избирательно, заключается в том, что они содержат гладкие мышцы . [42] Степень сокращения D4Z4 коррелирует с выраженностью извитости артериол. [42] Было высказано предположение, что ретинопатия обусловлена индуцированной белком DUX4 модуляцией оси CXCR4 – SDF1 , которая играет роль в морфологии клеток кончика эндотелия и ветвлении сосудов. [42]
Диагностика
[ редактировать ]
Во многих случаях ЛЛПД можно предположительно диагностировать на основании признаков, симптомов и/или негенетических медицинских тестов, особенно если у родственника первой линии генетически подтверждено ЛЛПД. [10] Генетическое тестирование может поставить окончательный диагноз. [4] При отсутствии установленного семейного анамнеза ЛЛПД постановка диагноза может быть затруднена из-за вариабельности проявлений ЛЛПД. [86]
Генетическое тестирование
[ редактировать ]Генетическое тестирование является золотым стандартом диагностики ЛЛПД, поскольку это наиболее чувствительный и специфичный доступный тест. [2] Обычно сначала проверяется FSHD1. [2] Укороченная длина массива D4Z4 ( длина EcoRI от 10 до 38 кб) с соседним аллелем 4qA поддерживает FSHD1. [2] Если FSHD1 отсутствует, обычно следующим тестируется FSHD2 путем оценки метилирования 4q35. [2] Для диагностики достаточно низкого метилирования (менее 20%) в контексте аллеля 4qA. [2] Конкретную мутацию, обычно одну из различных мутаций SMCHD1, можно идентифицировать с помощью секвенирования следующего поколения (NGS). [87]
Оценка длины D4Z4
[ редактировать ]Измерение длины D4Z4 технически сложно из-за массива повторов D4Z4, состоящего из длинных повторяющихся элементов. [88] Например, NGS бесполезен для оценки длины D4Z4, поскольку он разбивает ДНК на фрагменты перед их считыванием, и неясно, из какого повтора D4Z4 произошел каждый секвенированный фрагмент. [4] В 2020 году стало доступно оптическое картирование для измерения длины массива D4Z4, которое является более точным и менее трудоемким, чем Саузерн-блоттинг. [89] Молекулярное расчесывание также доступно для оценки длины массива D4Z4. [90] Эти методы, которые физически измеряют размер массива повторов D4Z4, требуют специально приготовленной высококачественной и высокомолекулярной геномной ДНК (гДНК) из сыворотки, что увеличивает стоимость и снижает доступность тестирования. [91]
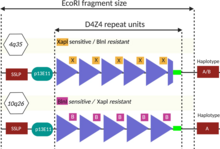
Анализ полиморфизма длин рестрикционных фрагментов (ПДРФ) был первым разработанным генетическим тестом, который до сих пор используется по состоянию на 2020 год, хотя его постепенно вытесняют новые методы. Он включает в себя нарезку ДНК рестрикционными ферментами и сортировку полученных рестрикционных фрагментов по размеру с использованием Саузерн-блоттинга . ферменты рестрикции Eco RI и Bln Обычно используются I. Eco RI изолирует массивы повторов 4q и 10q, а Bln I делит последовательность 10q на небольшие части, позволяя различить 4q. [4] [51] Рестрикционный фрагмент EcoRI состоит из трех частей: 1) проксимальной части размером 5,7 т.п.н., 2) центрального массива повторов D4Z4 переменного размера и 3) дистальной части, обычно размером 1,25 т.п.н. [92] Проксимальная часть имеет последовательность ДНК, окрашиваемую зондом p13E-11, который обычно используется для визуализации фрагмента EcoRI во время Саузерн-блоттинга. [51] Название «p13E-11» отражает то, что это субклон последовательности ДНК, обозначенной как космида 13E во время проекта генома человека. [93] [94] Учитывая, что каждый повтор D4Z4 имеет размер 3,3 т.п.н., а фрагмент EcoRI содержит около 5 т.п.н. ДНК, не входящей в состав массива повторов D4Z4, можно подсчитать количество единиц D4Z4. [78]
- Повторы D4Z4 = ( длина Эко RI - 5)/3,3
Иногда 4q или 10q имеют комбинацию повторов, подобных D4Z4 и D4Z4, из-за обмена ДНК между 4q и 10q, что может давать ошибочные результаты, требующие более детального изучения. [51] Иногда делеции массива повторов D4Z4 могут включать сайт связывания p13E-11, что требует использования альтернативных зондов. [51] [95]
Оценка статуса метилирования
[ редактировать ]Статус метилирования 4q35 традиционно оценивается после отрицательного результата теста на ЛЛПД1. Переваривание чувствительным к метилированию ферментом рестрикции (MSRE), показывающее гипометилирование, долгое время считалось диагностическим признаком ЛЛПД2. [91] Другие анализы метилирования были предложены или использованы в исследовательских целях, включая иммунопреципитацию метилированной ДНК и бисульфитное секвенирование , но обычно не используются в клинической практике. [96] [91] Бисульфитное секвенирование, если оно будет подтверждено, будет ценным, поскольку оно позволит использовать источники ДНК более низкого качества, например, обнаруженные в слюне. [91]
Вспомогательное тестирование
[ редактировать ]
Другие тесты могут подтвердить диагноз ЛЛПД, хотя все они менее чувствительны и менее специфичны, чем генетическое тестирование. [97] [4] Тем не менее, они могут исключить схожие условия. [14]
- Уровень креатинкиназы (КК) в крови часто определяют при подозрении на повреждение мышц. СК — это фермент , обнаруженный в мышцах, который попадает в кровь при повреждении мышц. При ЛЛПД уровень КК от нормального до слегка повышенного, [2] никогда не превышать в пять раз верхнюю границу нормы. [4]
- Электромиограмма (ЭМГ) измеряет электрическую активность в мышцах. ЭМГ может выявить неспецифические признаки повреждения мышц или раздражительности. [2]
- Скорость нервной проводимости (NCV) измеряет скорость распространения сигналов по нерву. Нервные сигналы измеряются с помощью поверхностных электродов (аналогичных тем, которые используются для электрокардиограммы) или игольчатых электродов. ЛЛПД не влияет на скорость нервной проводимости; аномальная нервная проводимость указывает на невропатию , а не на миопатию . [ нужна ссылка ]
- Биопсия мышц — это хирургическое удаление и исследование небольшого участка мышцы, обычно из руки или ноги. микроскопия и различные биохимические Для исследования используются тесты. Данные о ЛЛПД неспецифичны, например, наличие лейкоцитов или изменение размера мышечных волокон. Этот тест назначается редко. [2]
- Магнитно-резонансная томография (МРТ) чувствительна для обнаружения повреждений мышц даже в легких случаях. Т1-взвешенная МРТ позволяет визуализировать жировую инфильтрацию мышц, а Т2-взвешенная МРТ позволяет визуализировать мышечный отек. [ нужна ссылка ] МРТ может помочь дифференцировать ЛЛПД от других мышечных заболеваний на основе структуры задействованных мышц, направляя генетическое тестирование. [37] [38]
- Ультразвук (УЗИ) можно использовать для оценки мышц. [98] УЗИ эффективно при выявлении жировой инфильтрации или рубцевания мышц вследствие ЛЛПД, что проявляется повышенной эхогенностью (более яркой на ультразвуковом изображении). [98] Преимущества УЗИ по сравнению с МРТ или компьютерной томографией включают низкую стоимость и отсутствие радиационного воздействия. Однако при УЗИ хуже оцениваются более глубокие мышцы, а результаты ультразвукового исследования, как правило, в большей степени зависят от оператора. [98]
- Компьютерная томография (КТ) обычно не используется для визуализации мышц из-за значительного радиационного воздействия и плохой характеристики мягких тканей (т. е. частей тела, кроме костей). [98] Однако мышцы, случайно визуализированные с помощью КТ, могут выглядеть атрофированными и иметь меньшую плотность, поскольку жир менее плотен, чем мышцы. [98] КТ имеет ограниченную способность обнаруживать воспалительные изменения в мышцах, которые может обнаружить МРТ. [98]
Дифференциальный диагноз
[ редактировать ]В дифференциальный диагноз ЛЛПД включают поясно- конечностную мышечную дистрофию (особенно кальпаинопатию ), [2] лопаточно-перонеальная миопатия, [99] митохондриальная миопатия , [2] болезнь Помпе , [2] и полимиозит . [2] Кальпаинопатия и лопаточно-перонеальная миопатия, как и ЛЛПД, проявляются крыльями лопатки. [99] Признаками, указывающими на ЛЛПД, являются слабость лицевых мышц, асимметричная слабость и отсутствие эффекта от иммуносупрессивных препаратов. [2] Признаками, позволяющими поставить альтернативный диагноз, являются контрактуры , дыхательная недостаточность, слабость мышц, контролирующих движение глаз, а также слабость языка или горла . [14]
Управление
[ редактировать ]Ни одно фармакологическое лечение не доказало значительного замедления прогрессирования слабости или значительного улучшения силы. [100] [2] [10]
Скрининг и мониторинг осложнений
[ редактировать ]Американская академия неврологии (AAN) рекомендует несколько медицинских тестов для выявления осложнений ЛЛПД. [100] с расширением глаз Обследование глаз для выявления аномалий сетчатки рекомендуется тем, у кого впервые диагностирован ЛЛПД; тем, у кого есть большие делеции D4Z4, рекомендуется ежегодно проходить обследование у специалиста по сетчатке. [101] [2] Проверка слуха рекомендуется людям с ЛЛПД с ранним началом перед поступлением в школу или любому другому человеку с ЛЛПД с симптомами потери слуха. [101] [2] Тестирование функции легких (PFT) рекомендуется пациентам с впервые поставленным диагнозом, чтобы установить исходную функцию легких. [2] и периодически для лиц с симптомами или рисками легочной недостаточности. [101] [2] Рутинный скрининг заболеваний сердца, например, с помощью электрокардиограммы (ЭКГ) или эхокардиограммы (эхо), считается ненужным для лиц без симптомов сердечно-сосудистых заболеваний. [100]
Физиотерапия и трудотерапия
[ редактировать ]аэробные упражнения уменьшают хроническую усталость и замедляют жировую инфильтрацию мышц при ЛЛПД. Было показано, что [102] [103] Физическая активность в целом может замедлить прогрессирование заболевания ног. [10] AAN рекомендует людям с ЛЛПД заниматься аэробными упражнениями низкой интенсивности, чтобы повысить уровень энергии, здоровье мышц и костей. [2] Силовые тренировки средней интенсивности, по-видимому, не приносят вреда, хотя не было доказано, что они приносят пользу. [104] Физиотерапия может устранить конкретные симптомы; стандартизированного протокола для ЛЛПД не существует. По неофициальным данным, правильно наложенная кинезиологическая лента может уменьшить боль. [105] Трудотерапию можно использовать для обучения повседневной деятельности (ADL) и для помощи в адаптации к новым вспомогательным устройствам . когнитивно-поведенческая терапия (КПТ) снижает хроническую усталость при ЛЛПД, а также замедляет жировую инфильтрацию мышц, если она направлена на повышение повседневной активности. Было показано, что [102] [103]
Брекеты часто используются для устранения мышечной слабости. Фиксация лопатки может улучшить положение лопатки, что улучшает функцию плеча, хотя ее часто считают неэффективной или непрактичной. [106] Ортезы голеностопного сустава улучшают ходьбу, баланс и качество жизни. [107]
Фармакологический менеджмент
[ редактировать ]Ни один фармацевтический препарат не доказал свою эффективность в изменении течения заболевания. [100] Хотя некоторые фармацевтические препараты показали улучшение мышечной массы в ограниченных отношениях, они не улучшили качество жизни, и AAN не рекомендует их использовать при ЛЛПД. [100]
Реконструктивная хирургия
[ редактировать ]Крылья лопатки поддаются хирургической коррекции, а именно оперативной фиксации лопатки. Фиксация лопатки – это ограничение и стабилизация положения лопатки, ее более близкое прилегание к грудной клетке и уменьшение крылатости. Чаще всего сообщается об абсолютном ограничении движения лопатки за счет фиксации лопатки к ребрам. [108] Эта процедура часто включает в себя сращение костей, называемое артродезом , между лопаткой и ребрами. Названия этого явления включают лопаточно-грудной спондилодез, спондилодез лопатки и лопатодез. Эта процедура увеличивает активный диапазон движений руки , улучшает функцию руки, уменьшает боль и улучшает косметический вид. [109] [110] Активный диапазон движений руки увеличивается больше всего при тяжелом расправлении лопатки с непораженной дельтовидной мышцей ; [11] однако пассивный диапазон движений уменьшается. Другими словами, пациент приобретает способность медленно поднимать руки на 90+ градусов, но теряет способность «подбрасывать» руку на полные 180 градусов. [2] AAN утверждает, что фиксацию лопатки можно предлагать с осторожностью некоторым пациентам, сопоставив эти преимущества с неблагоприятными последствиями хирургического вмешательства и длительной иммобилизации. [100] [10]
Другой формой оперативной фиксации лопатки является скапулопексия. «Scapulo-» относится к кости лопатки, а «-pexy» происходит от греческого корня «связывать». Некоторые варианты скапулопексии дают по существу тот же результат, что и спондилодез лопаточно-грудного сустава, но вместо сращения костей лопатка фиксируется к ребрам только с помощью проволоки, сухожильных трансплантатов или другого материала. Некоторые варианты скапулопексии не полностью ограничивают движение лопатки, например, привязывают лопатку к ребрам, позвонкам или другой лопатке. [108] [111] Скапулопексия считается более консервативной, чем лопаточно-грудной спондилодез, с меньшим временем восстановления и меньшим влиянием на дыхание. [108] Однако они также кажутся более подверженными долгосрочным неудачам. [108] Другой формой фиксации лопатки, хотя и не часто применяемой при ЛЛПД, является перенос сухожилий , который включает в себя хирургическую перестановку мест прикрепления мышц к кости. [108] [112] [113] Примеры включают перенос большой грудной мышцы и процедуру Идена-Ланге .
Описаны различные другие хирургические реконструкции. Золотые имплантаты верхнего века используются для тех, кто не может закрыть глаза. [114] Опущенную нижнюю губу лечат пластической хирургией. [115] Способность улыбаться теоретически может быть восстановлена с помощью трансплантации сухожилия с использованием доноров, таких как часть височной мышцы , хотя доказательства в отношении ЛЛПД отсутствуют. [116] В некоторых случаях опущение стопы можно исправить хирургическим путем с помощью переноса сухожилия, при котором задняя большеберцовая мышца перепрофилируется в переднюю большеберцовую мышцу , вариант этого метода называется процедурой уздечки. [117] [118] [105] Тяжелый сколиоз, вызванный ЛЛПД, можно исправить с помощью спондилодеза ; однако, поскольку сколиоз может быть компенсаторным изменением в ответ на мышечную слабость, коррекция выравнивания позвоночника может привести к дальнейшему нарушению мышечной функции.
- Управление лопаточным крылом
 Кинезиотейп накладывается на лопатки.
Кинезиотейп накладывается на лопатки. Тканевый бандаж, удерживающий лопатки в отведенном положении для уменьшения симптомов плеча, таких как боль в ключицах.
Тканевый бандаж, удерживающий лопатки в отведенном положении для уменьшения симптомов плеча, таких как боль в ключицах. Скапулопексия «лопатка-лопатка» до и после операции. Лопатки связаны вместе в втянутом положении с помощью трансплантата ахиллова сухожилия , что в данном случае позволило различить большие ромбовидные мышцы.
Скапулопексия «лопатка-лопатка» до и после операции. Лопатки связаны вместе в втянутом положении с помощью трансплантата ахиллова сухожилия , что в данном случае позволило различить большие ромбовидные мышцы.



Прогноз
[ редактировать ]Генетика частично предсказывает прогноз. [100] Те, у кого есть большие делеции повторов D4Z4 (с размером оставшегося массива повторов D4Z4 10–20 т.п.н. или 1–4 повторов), с большей вероятностью будут иметь тяжелое и раннее заболевание, а также немышечные симптомы. [100] Те, у кого есть генетические мутации как ЛЛПД1, так и ЛЛПД2, с большей вероятностью будут иметь тяжелое заболевание. [74] Также было замечено, что укорочение D4Z4 меньше и проявления заболевания мягче при наличии выраженного семейного анамнеза, в отличие от новой мутации. [119] В некоторых больших семьях у 30% людей с мутацией не было заметных симптомов, а у 30% людей с симптомами болезнь не прогрессировала дальше слабости лица и плеч. [24] У женщин, как правило, симптомы развиваются в более позднем возрасте, и заболевание протекает менее тяжело. [113]
Остальные вариации течения заболевания объясняются неизвестными факторами окружающей среды. Одно исследование показало, что курение табака или употребление алкоголя, являющиеся общими факторами риска других заболеваний, не ухудшают течение заболевания. [120]
Беременность
[ редактировать ]Исходы беременности в целом хорошие у матерей с ЛЛПД; нет никакой разницы в частоте преждевременных родов , частоте выкидышей и исходах у младенцев. [121] Однако слабость может увеличить потребность в вспомогательных родах . [121]
В одном обзоре было обнаружено, что слабость усиливается без выздоровления у 12% матерей с ЛЛПД во время беременности, хотя это может быть связано с увеличением веса или ухудшением физической формы . [121]
Эпидемиология
[ редактировать ]Распространенность . ЛЛПД колеблется от 1 на 8333 до 1 на 15 000 [2] В Нидерландах сообщается о распространенности 1 на 8 333, если учитывать невыявленные случаи. [122] Распространенность в Соединенных Штатах обычно оценивается как 1 на 15 000. [15]
После того, как в 1992 году стало возможным генетическое тестирование, средняя распространенность составила около 1 на 20 000, что является значительным увеличением по сравнению с периодом до 1992 года. [123] [24] [122] Тем не менее, 1 из 20 000, вероятно, является заниженной оценкой, поскольку многие люди с ЛЛПД имеют легкие симптомы и никогда не диагностируются, или они являются братьями и сестрами больных людей и никогда не обращаются за окончательным диагнозом. [122]
Не было доказано, что раса и этническая принадлежность влияют на частоту или тяжесть ЛЛПД. [15]
Хотя при наследовании ЛЛПД не наблюдается предрасположенности к биологическому полу, заболевание реже проявляется у женщин, и даже когда оно манифестирует у женщин, они в среднем страдают менее тяжело, чем мужчины. [15] эстроген Предполагается, что является защитным фактором, объясняющим это несоответствие. Одно исследование показало, что эстроген снижает активность DUX4 . [124] Однако другое исследование не выявило связи между тяжестью заболевания и воздействием эстрогена у женщин в течение жизни. В том же исследовании было обнаружено, что прогрессирование заболевания не различалось в периоды гормональных изменений, таких как менархе , беременность и менопауза . [125]
История
[ редактировать ]Первое описание человека с ЛЛПД в медицинской литературе появляется в отчете о вскрытии Жана Крювейье в 1852 году. [16] [17] В 1868 году Дюшенн опубликовал свою основополагающую работу о мышечной дистрофии Дюшенна , частью которой . было описание ЛЛПД [126] [17] Сначала в 1874 году, затем в более часто цитируемой публикации в 1884 году и снова с фотографиями в 1885 году французские врачи Луи Ландузи и Жозеф Дежерин опубликовали подробности болезни, признав ее отдельной клинической единицей, и поэтому ЛЛПД иногда называют как болезнь Ландузи-Джерина. [18] [17] В своей статье 1886 года Ландузи и Дежерин обратили внимание на семейную природу заболевания и упомянули, что у родственников, которых они исследовали, было затронуто четыре поколения. [127] Формального определения клинических особенностей ЛЛПД не было до 1952 года, когда была изучена большая семья из штата Юта с ЛЛПД. Начиная примерно с 1980 года растущий интерес к ЛЛПД привел к лучшему пониманию большой изменчивости заболевания и растущему пониманию генетических и патофизиологических сложностей. К концу 1990-х годов исследователи наконец начали понимать области хромосомы 4, связанные с ЛЛПД. [53]
С момента публикации объединяющей теории в 2010 году исследователи продолжали совершенствовать свое понимание DUX4 . С ростом доверия к этой работе исследователи в 2014 году впервые предложили консенсусную точку зрения на патофизиологию заболевания и потенциальные подходы к терапевтическому вмешательству, основанные на этой модели. [27]
Альтернативные и исторические названия ЛЛПД включают следующее:
- лице-лопаточно-плечевая болезнь [23]
- лице-плече-лопаточный [ нужна ссылка ]
- Болезнь Ландузи-Дежерина [23] (или «синдром» [127] или «тип мышечной дистрофии» [23] )
- Синдром Эрба-Ландузи-Дежерина [ нужна ссылка ]
Хронология важных генетических исследований, связанных с ЛЛПД
[ редактировать ]| 1861 | Человек с мышечной дистрофией, изображенный Дюшенном. Судя по задействованным мышцам, у этого человека мог быть ЛЛПД.  |
| 1884 | Ландузи и Дежерин описывают форму прогрессирующей мышечной атрофии у детей с характерным поражением лицевых мышц, отличающуюся от псевдогипертрофической (МД Дюшенна) и атрофии спинальных мышц у взрослых. [128] Два брата с ЛЛПД, за ними следуют Ландузи и Дежерин. Фотография одного брата в возрасте 21 года. Правая лопатка вытянута, ротирована вниз и смещена латерально. Рисунок другого брата в 17 лет. Виден поясничный гиперлордоз. Плечо и грудные мышцы кажутся атрофированными. |
| 1886 | Ландузи и Дежерин описывают прогрессирующую мышечную атрофию лопаточно-плечевого типа. [129] |
| 1950 | Тайлер и Стивенс изучили 1249 человек из одного рода с ЛЛПД, происходящим от одного предка, и описали типичный менделевский образец наследования с полной пенетрантностью и очень вариабельным проявлением. Введен термин лице-лопаточно-плечевая дистрофия . [130] |
| 1982 | Падберг представил первые исследования сцепления для определения генетического локуса ЛЛПД в своей основополагающей диссертации «Лице-лопаточно-плечевая болезнь». [23] |
| 1987 | полная последовательность гена дистрофина ( МД Дюшенна ). Определена [131] |
| 1991 | Генетический дефект при ЛЛПД связан с участком (4q35) вблизи кончика длинного плеча хромосомы 4 . [132] |
| 1992 | ЛЛПД, как в семейных, так и в случаях de novo Обнаружено, что , связан с событием рекомбинации, которое уменьшает размер фрагмента 4q Eco R1 до <28 т.п.н. (обычно 50–300 т.п.н.). [93] |
| 1993 | Обнаружено, что фрагменты 4q Eco R1 содержат тандемное расположение нескольких единиц размером 3,3 т.п.н. (D4Z4), а FSHD связан с наличием <11 единиц D4Z4. [92] Исследование семи семей с ЛЛПД выявило доказательства генетической гетерогенности ЛЛПД. [133] |
| 1994 | Гетерохроматическая позиционно структура 4q35 признана фактором, который может влиять на экспрессию ЛЛПД, возможно, посредством -эффектного разнообразия . [134] Секвенирование ДНК внутри единиц D4Z4 показывает, что они содержат открытую рамку считывания, соответствующую двум доменам гомеобокса , но исследователи приходят к выводу, что D4Z4 вряд ли будет кодировать функциональный транскрипт. [134] [135] |
| 1995 | Термины ЛЛПД1А и ЛЛПД1В введены для описания 4q-связанных и несвязанных с 4q форм заболевания. [136] |
| 1996 | Ген 1 региона FSHD ( FRG1 ) обнаружен в 100 т.п.н. проксимальнее D4Z4. [137] |
| 1998 | монозиготные близнецы с совершенно разными клиническими проявлениями ЛЛПД. Описаны [20] |
| 1999 | Полное секвенирование единиц D4Z4 4q35 выявляет область промотора, расположенную на расстоянии 149 п.о. 5' от открытой рамки считывания для двух доменов гомеобокса, что указывает на ген, который кодирует белок, состоящий из 391 аминокислоты (позже исправленный до 424 аминокислотных остатков). [138] ), получивший название DUX4 . [139] |
| 2001 | Исследователи оценили состояние метилирования ( гетерохроматин более сильно метилирован, чем эухроматин ) ДНК в 4q35 D4Z4. Исследование Sma I, Mlu I, Sac II и Eag I рестрикционных фрагментов из нескольких типов клеток, включая скелетные мышцы, не выявило доказательств гипометилирования в клетках пациентов с ЛЛПД1 по сравнению с D4Z4 из непораженных контрольных клеток или по сравнению с гомологичными сайтами D4Z4 на хромосома 10 . Однако во всех случаях D4Z4 из сперматозоидов был гипометилирован по сравнению с D4Z4 из соматических тканей. [140] |
| 2002 | Обнаружено, что полиморфный сегмент длиной 10 т.п.н. непосредственно дистальнее D4Z4 существует в двух аллельных формах, обозначенных 4qA и 4qB. FSHD1 связан исключительно с аллелем 4qA. [141] Три гена (FRG1, FRG2 , ANT1 ), расположенные в области центромерно по отношению к D4Z4 на хромосоме 4, обнаружены в изолированных мышечных клетках людей с ЛЛПД на уровнях, в 10–60 раз превышающих нормальные, что указывает на связь между сокращениями D4Z4 и измененной экспрессией Гены 4q35. [142] |
| 2003 | Дальнейшее исследование метилирования ДНК в различных рестрикционных фрагментах 4q35 D4Z4 ( BsaA I и Fse I) показало значительное гипометилирование в обоих сайтах у людей с ЛЛПД1, носителей гена, не экспрессирующего ЛЛПД, и людей с фенотипическим ЛЛПД по сравнению с незатронутым контролем. [143] |
| 2004 | Сокращение региона D4Z4 на аллеле 4qB до <38 т.п.н. не вызывает ЛЛПД. [144] |
| 2006 | Показано, что у трансгенных мышей со сверхэкспрессией FRG1 развивается тяжелая миопатия. [145] |
| 2007 | Обнаружено, что открытая рамка считывания DUX4 сохраняется в геноме приматов более 100 миллионов лет, что подтверждает вероятность того, что она кодирует необходимый белок. [146] Исследователи идентифицируют мРНК DUX4 в первичных миобластах ЛЛПД и идентифицируют в клетках, трансфицированных D4Z4, белок DUX4, сверхэкспрессия которого индуцирует гибель клеток. [138] и белка DUX4 мРНК Сообщается, что экспрессия увеличивается в миобластах у пациентов с ЛЛПД по сравнению с незатронутой группой контроля. Стабильная мРНК DUX4 транскрибируется только с самой дистальной единицы D4Z4, которая использует интрон и сигнал полиаденилирования, обеспечиваемый фланкирующей областью pLAM. Белок DUX4 идентифицирован как фактор транскрипции, и данные свидетельствуют о том, что сверхэкспрессия DUX4 связана с увеличением целевого парного гомеодоменного транскрипционного фактора 1 ( PITX1 ). [147] |
| 2009 | Термины FSHD1 и FSHD2 введены для описания генетических форм, связанных с делецией D4Z4, и не связанных с делецией D4Z4, соответственно. При ЛЛПД1 гипометилирование ограничивается коротким аллелем 4q, тогда как ЛЛПД2 характеризуется гипометилированием как 4q, так и обоих аллелей 10q. [148] что сплайсинг и расщепление терминального (наиболее теломерного ) транскрипта 4q D4Z4 DUX4 в первичных миобластах и фибробластах пациентов с ЛЛПД приводит к образованию множественных РНК, включая малые некодирующие РНК , антисмысловые РНК и кэпированные мРНК в качестве новых кандидатов для патофизиологии Обнаружено , ФСДД. [149] Предложенный механизм DBE-T (транскрипт регуляторного элемента D4Z4), приводящий к дерепрессии генов 4q35. [62] |
| 2010 | Установлена объединяющая генетическая модель ЛЛПД: сокращения D4Z4 вызывают ЛЛПД только в контексте аллели 4qA из-за стабилизации транскрипта РНК DUX4 , что обеспечивает экспрессию DUX4 . [8] Несколько организаций, включая The New York Times, отметили это исследование. [150] [151] Фрэнсис Коллинз , курировавший первое секвенирование генома человека в Национальном институте здравоохранения, заявил: [150]
Дэниел Перес, соучредитель Общества FSHD, приветствовал новые результаты, сказав: [151]
MDA заявило, что: [ нужна ссылка ]
Один из соавторов доклада, Сильвер ван дер Маарель из Лейденского университета, заявил, что [ нужна ссылка ]
DUX4 активно транскрибируется в биоптатах скелетных мышц и первичных миобластах. Клетки, пораженные ЛЛПД, продуцируют полноразмерный транскрипт, DUX4-fl, тогда как альтернативный сплайсинг у здоровых людей приводит к образованию более короткого, 3'-усеченного транскрипта (DUX4-s). Низкая общая экспрессия обоих транскриптов в мышцах объясняется относительно высокой экспрессией в небольшом количестве ядер (~ 1 на 1000). Более высокие уровни экспрессии DUX4 в семенниках человека (примерно в 100 раз выше, чем в скелетных мышцах) предполагают роль DUX4 в развитии человека. Показано, что более высокие уровни DUX4-s (по сравнению с DUX4-fl) коррелируют с большей степенью метилирования DUX-4 H3K9me3. [7] |
| 2012 | Некоторые случаи ЛЛПД2 связаны с мутациями в гене SMCHD1 на хромосоме 18 , и установлено генетическое/механистическое пересечение ЛЛПД1 и ЛЛПД2. [68] Распространенность ЛЛПД-подобных делеций D4Z4 на пермиссивных аллелях значительно выше, чем распространенность ЛЛПД в общей популяции, что бросает вызов критериям молекулярной диагностики ЛЛПД. [152] При экспрессии в первичных миобластах DUX4-fl действовал как активатор транскрипции, вызывая >3-кратное изменение экспрессии 710 генов. [153] Последующее исследование с использованием большего количества образцов выявило, что экспрессия DUX4-fl в миогенных клетках и мышечной ткани непораженных родственников пациентов с ЛЛПД сама по себе недостаточна для того, чтобы вызвать патологию, и что дополнительные модификаторы являются детерминантами прогрессирования заболевания. [154] |
| 2013 | Показано, что мутации в SMCHD1 увеличивают тяжесть ЛЛПД1. [74] Было обнаружено, что трансгенные мыши , несущие массивы D4Z4 от аллеля FSHD1 (с 2,5 единицами D4Z4), хотя и лишены очевидного FSHD-подобного фенотипа скелетных мышц, воспроизводят важные паттерны генетической экспрессии и эпигенетические особенности FSHD. [155] |
| 2014 | DUX4-fl и нижестоящие гены-мишени экспрессируются в биоптатах скелетных мышц и полученных из биопсии клетках плодов с FSHD-подобными массивами D4Z4, что указывает на то, что молекулярные маркеры FSHD уже экспрессируются во время развития плода. [156] Исследователи «рассматривают, как вклад многих лабораторий на протяжении многих лет привел к пониманию фундаментально нового механизма заболеваний человека» и формулируют, как объединяющая генетическая модель и последующие исследования представляют собой «поворотную точку в исследованиях ЛЛПД, переходящую от открытий». от исследований, ориентированных на трансляционные исследования, направленные на разработку методов лечения, основанных на надежной модели патофизиологии заболеваний». Они описывают консенсусный механизм патофизиологии ЛЛПД как «неэффективную опосредованную повторами эпигенетическую репрессию массива макросателлитных повторов D4Z4 на хромосоме 4, что приводит к разнообразной экспрессии ретрогена DUX4 , кодирующего фактор транскрипции с двойным гомеобоксом, в скелетных мышцах. " [27] |
| 2020 | Опубликован отчет «Голос пациента», документирующий влияние ЛЛПД на повседневную жизнь, о котором рассказали около 400 пациентов во время совещания FDA по разработке лекарств, ориентированных на пациентов, которое проводилось под внешним руководством FDA, которое состоялось 29 июня 2020 года. [32] [30] [31] [33] |


Прошлые фармацевтические разработки
[ редактировать ]Ранние испытания препарата, до того как был открыт патогенез DUX4 , были нецелевыми и в основном безуспешными. [19] Соединения испытывались с целью увеличения мышечной массы, уменьшения воспаления или рассмотрения предварительных теорий механизма заболевания. [19] Следующие препараты не показали эффективности:
- Преднизолон , стероид, основанный на его противовоспалительных свойствах и терапевтическом эффекте при мышечной дистрофии Дюшенна. [157]
- Пероральный альбутерол , β2-агонист , на основании его анаболических свойств. Хотя это улучшило мышечную массу и некоторые показатели силы у людей с ЛЛПД, оно не улучшило общую силу или функции. [158] [159] [160] Интересно, что позже было показано, что агонисты β2 снижают экспрессию DUX4. [161]
- Дилтиазем , блокатор кальциевых каналов , на основе неофициальных сообщений о пользе и теории о том, что нарушение регуляции кальция играет определенную роль в гибели мышечных клеток. [162]
- MYO-029 ( стамулумаб ), антитело, ингибирующее миостатин , было разработано для стимулирования мышечного роста. Миостатин – это белок, который подавляет рост мышечной ткани. [163]
- ACE-083, ингибитор TGF-β, был разработан для стимулирования мышечного роста. [164]
Общество и культура
[ редактировать ]СМИ
[ редактировать ]- В Amazon Video сериале «Человек в высоком замке » сыну обергруппенфюрера Джона Смита, Томасу, поставлен диагноз синдром Ландузи-Дежерина. [165]
- В биографии «Стюарт: Жизнь наоборот» главный герой страдал мышечной дистрофией. [166] предположительно ФЛСД. [ нужна ссылка ]
- «Хорошие плохие дела» — независимый фильм о предпринимателе, страдающем ЛЛПД, и его пути трансформации, самопринятия и открытий. Премьера фильма состоялась на кинофестивале Slamdance 2024. [167] [168]
- «Потерянный голос» — короткометражный фильм с участием Тристрама Ингэма, живущего с ЛЛПД. Apple Ингэм использовал функцию «Персональный голос» , чтобы воссоздать свой голос и использовать его для чтения детской книги « Потерянный голос» к Международному дню людей с ограниченными возможностями в 2023 году. [169]
Пациенты и исследовательские организации
[ редактировать ]- Общество FSHD (до 2019 года называлось «Общество FSH») [170] была основана в 1991 году на Восточном побережье двумя людьми с ЛЛПД, Дэниелом Пересом и Стивеном Якобсеном. [171] [172] Общество ЛЛПД утверждает, что выступило за стандартизацию названия заболевания лице-лопаточно-плечевая мышечная дистрофия и его аббревиатуры ЛЛПД . [170] Общество FSHD утверждает, что оно собрало финансирование для стартовых грантов на исследования FSHD и стало соавтором Закона MD-CARE 2001 года, который предоставил федеральное финансирование для всех мышечных дистрофий. [171] [172] Общество ЛЛПД превратилось в крупнейшую в мире массовую организацию, выступающую за обучение пациентов, а также научные и медицинские исследования в области ЛЛПД. [173] [174] Одним из известных представителей Общества FSHD был Макс Адлер , актер телесериала Glee . [175]
- Friends of FSH Research — это исследовательская некоммерческая организация, основанная в 2004 году Терри и Риком Колеллой из Кирклэнда, штат Вашингтон, после того, как у их сына был диагностирован ЛЛПД. [176] [177] [178] [179]
- был Глобальный исследовательский фонд FSHD основан в 2007 году Биллом Моссом в Австралии, бывшим руководителем Macquarie Bank, пострадавшим от FSHD. [180] [181] [182] В настоящее время режиссером является дочь Мосса. [181] В 2017 году он был назван австралийской благотворительной организацией года. [181] По состоянию на 2018 год это крупнейший спонсор медицинских исследований ЛЛПД за пределами США. [182]
- FSHD EUROPE была основана в 2010 году. [183] Охватывая несколько стран Европы, он запустил Европейскую сеть судебных разбирательств. [183]
Известные случаи
[ редактировать ]- Чип Уилсон — канадский миллиардер и основатель компании Lululemon . Он пообещал выделить 100 миллионов канадских долларов на исследования в рамках проекта «Solve FSHD». [184]
- Крис Каррино — радиоголос команды « Бруклин Нетс» . Он основал Фонд Криса Каррино по ЛЛПД, ориентированный на финансирование исследований. [185]
- Мэдисон Феррис — американская актриса с ЛЛПД, первая инвалид-колясочник, сыгравшая главную роль на Бродвее . [186] [187]
- Морган Хоффманн — американский профессиональный игрок в гольф. Он основал Фонд Моргана Хоффмана, ориентированный на финансирование исследований. [188]
- Арнольд Голд (1925–2024) был детским неврологом, основателем Фонда Арнольда П. Голда и создателем церемоний белых халатов . Голд сосредоточился на заботе, ориентированной на пациента, и гуманизме . [189]
Направления исследований
[ редактировать ]Фармацевтические разработки
[ редактировать ]Достигнув консенсуса по патофизиологии ЛЛПД в 2014 году, исследователи предложили четыре подхода к терапевтическому вмешательству: [27]
- усилить эпигенетическую репрессию D4Z4
- нацеливать мРНК DUX4, включая изменение сплайсинга или полиаденилирования;
- блокировать активность белка DUX4
- ингибировать DUX4-индуцированный процесс или процессы, которые приводят к патологии.
Маломолекулярные препараты
Большинство лекарств, используемых в медицине, представляют собой « маломолекулярные лекарства », в отличие от биологических медицинских продуктов, которые включают белки, вакцины и нуклеиновые кислоты. Маломолекулярные препараты обычно можно принимать внутрь, а не в виде инъекций.
- Лосмапимод , селективный ингибитор митоген-активируемых протеинкиназ p38α/β , был идентифицирован компанией Fulcrum Therapeutics как мощный супрессор DUX4 in vitro. [191] Результаты клинического исследования фазы IIb, опубликованные в июне 2021 года, показали статистически значимое замедление ухудшения мышечной функции. Ожидаются дальнейшие судебные разбирательства. [192] [193]
- казеинкиназы 1 Голландская фармацевтическая компания Facio Therapies обнаружила, что ингибирование (CK1) подавляет экспрессию DUX4 и находится в стадии доклинической разработки. Facio Therapies утверждает, что ингибирование CK1 оставляет слияние мышечных трубочек нетронутым, в отличие от ингибиторов BET, ингибиторов p38 MAPK и агонистов β2 . [194] [195]
Генная терапия
Генная терапия – это введение нуклеотидов для лечения заболеваний. Несколько типов генной терапии для лечения ЛЛПД находятся на доклинической стадии разработки.
- В антисмысловой терапии используются антисмысловые олигонуклеотиды , которые связываются с информационной РНК DUX4, вызывая ее деградацию и предотвращая выработку белка DUX4. фосфородиамидат морфолино , олигонуклеотид, модифицированный для повышения его стабильности Было показано, что , избирательно снижает DUX4 и его эффекты; однако эти антисмысловые нуклеотиды обладают плохой способностью проникать в мышцы. [2]
- микроРНК (миРНК), направленные против DUX4, доставляемые вирусными векторами , уменьшают DUX4, защищают от мышечной патологии и предотвращают потерю силы захвата на моделях FSHD у мышей. Было показано, что [2] Компания Arrowhead Pharmacys разрабатывает терапевтический препарат с РНК-интерференцией против DUX4 под названием ARO-DUX4 и намерена подать заявку на получение разрешения регулирующих органов в третьем квартале 2021 года, чтобы начать клинические испытания. [196] [197] [198]
- Редактирование генома , постоянное изменение генетического кода, исследуется. В одном исследовании пытались использовать CRISPR-Cas9 для отключения сигнала полиаденилирования на лабораторных моделях, но не удалось показать терапевтические результаты. [199]
Потенциальные мишени для наркотиков
- Ингибирование пути гиалуроновой кислоты (ГК) является потенциальной терапией. Одно исследование показало, что многие DUX4 -индуцированные молекулярные патологии опосредованы передачей сигналов HA, а ингибирование биосинтеза HA с помощью 4-метилумбеллиферона предотвращает эти молекулярные патологии. [200]
- Показано, что ингибирование P300 подавляет вредные эффекты DUX4. [201]
- ингибиторы BET снижают экспрессию DUX4. Было показано, что [161]
- Антиоксиданты потенциально могут уменьшить последствия ЛЛПД. Одно исследование показало, что прием витамина С , витамина Е , глюконата цинка и селенометионина увеличивает выносливость и силу четырехглавых мышц, но не оказывает существенного улучшения на ходьбу. [202] Дальнейшее изучение оправдано. [2]
Результаты измерения
[ редактировать ]Способы измерения заболевания важны для изучения прогрессирования заболевания и оценки эффективности лекарств в клинических испытаниях.
- В Reachable Workspace используется система определения движения Kinect для сравнения диапазона досягаемости до и после терапевтического клинического исследования. [203]
- Качество жизни можно измерить с помощью опросников, таких как Индекс здоровья FSHD. [204] [2]
- То, как заболевание влияет на повседневную деятельность, можно измерить с помощью опросников, таких как построенная по методу FSHD-Rasch (FSHD-RODS). шкала общей инвалидности, [205] или комбинированный показатель результата FSHD (FSHD-COM). [206]
- Электроимпедансная миография изучается как способ измерения повреждения мышц. [2]
- МРТ мышц полезна для оценки всех мышц тела. Мышцы можно оценить по степени жировой инфильтрации. [2]
Ссылки
[ редактировать ]- ^ Перечисленные ниже источники различаются по произношению буквы «u» в слове «scapulo». Звук «долгий у» в безударном нефинальном слоге часто сокращается до шва и варьируется в зависимости от говорящего.
- «Лице-лопаточно-плечевой» . Словарь Merriam-Webster.com .
- «Лице-лопаточно-плечевой» . Медицинский словарь, Фарлекс и партнеры, 2009.
- ^ Перейти обратно: а б с д и ж г час я дж к л м н тот п д р с т в v В х и С аа аб и объявление но из в ах есть также и аль являюсь а к ап ак с как в В из хорошо Вагнер КР (декабрь 2019 г.). «Лице-лопаточно-плечевые мышечные дистрофии». КОНТИНУУМ: Непрерывное обучение в области неврологии . 25 (6): 1662–1681. дои : 10.1212/CON.0000000000000801 . ПМИД 31794465 . S2CID 208531681 .
- ^ Стедман Т. (1987). Новый мир Вебстера / Краткий медицинский словарь Стедмана (1-е изд.). Балтимор: Уильямс и Уилкинс. п. 230. ИСБН 0-13-948142-7 .
- ^ Перейти обратно: а б с д и ж г час я дж к л м н тот п д р с т в v В х и С аа аб и объявление но Мул К., Лассе С., Фёрманс Н.К., Падберг Г.В., Хорлингс К.Г., ван Энгелен Б.Г. (июнь 2016 г.). «Что в названии? Клинические особенности лице-лопаточно-плечевой мышечной дистрофии». Практическая неврология . 16 (3): 201–207. doi : 10.1136/practneurol-2015-001353 . ПМИД 26862222 . S2CID 4481678 .
- ^ Перейти обратно: а б Кумар В., Аббас А., Астер Дж., ред. (2018). Основная патология Роббинса (Десятое изд.). Филадельфия, Пенсильвания: Эльзевир. п. 844. ИСБН 978-0-323-35317-5 .
- ^ Де Яко А, Планета Е, Колуччо А, Верп С, Дюк Дж, Троно Д (июнь 2017 г.). «Факторы транскрипции семейства DUX регулируют активацию зиготического генома у плацентарных млекопитающих» . Природная генетика . 49 (6): 941–945. дои : 10.1038/ng.3858 . ПМК 5446900 . ПМИД 28459456 .
- ^ Перейти обратно: а б с Снайдер Л., Гэн Л.Н., Леммерс Р.Дж., Киба М., Уэр С.Б., Нельсон А.М., Тавил Р., Филиппова Г.Н., ван дер Маарел С.М., Тапскотт С.Дж., Миллер Д.Г. (28 октября 2010 г.). «Лице-лопаточно-плечевая дистрофия: неполное подавление ретротранспонированного гена» . ПЛОС Генетика . 6 (10): e1001181. дои : 10.1371/journal.pgen.1001181 . ПМЦ 2965761 . ПМИД 21060811 .
- ^ Перейти обратно: а б с д и ж г час я дж к Леммерс Р.Дж., ван дер Влит П.Дж., Клоостер Р., Саккони С., Каманьо П., Дауверсе Дж.Г., Снайдер Л., Страашейм К.Р., ван Оммен Г.Дж., Падберг Г.В., Миллер Д.Г., Тапскотт С.Дж., Тавил Р., Франц Р.Р., ван дер Маарель С.М. ( 19 августа 2010 г.). «Объединяющая генетическая модель лице-лопаточно-плечевой мышечной дистрофии» (PDF) . Наука . 329 (5999): 1650–3. Бибкод : 2010Sci...329.1650L . дои : 10.1126/science.1189044 . hdl : 1887/117104 . ПМЦ 4677822 . ПМИД 20724583 . Архивировано из оригинала (PDF) 5 июня 2014 г.
- ^ Перейти обратно: а б Леммерс Р.Дж., Вольгемут М., ван дер Гааг К.Дж. и др. (ноябрь 2007 г.). «Конкретные вариации последовательности в области 4q35 связаны с лице-лопаточно-плечевой мышечной дистрофией» . Являюсь. Дж. Хум. Жене . 81 (5): 884–94. дои : 10.1086/521986 . ПМК 2265642 . ПМИД 17924332 .
- ^ Перейти обратно: а б с д и ж г час я дж к л Мул К (1 декабря 2022 г.). «Лице-лопаточно-плечевая мышечная дистрофия». Континуум (Миннеаполис, Миннесота) . 28 (6): 1735–1751. дои : 10.1212/CON.0000000000001155 . ПМИД 36537978 . S2CID 254883066 .
- ^ Перейти обратно: а б Эрен И, Бирсел О, Озтоп Чакмак О, Аслангер А, Гюрсой Оздемир Ю, Эраслан С, Кайсерили Х, Офлазер П, Демирхан М (май 2020 г.). «Новая система стадирования инвалидности плечевого сустава при артродезе лопаточно-грудного отдела у пациентов с лице-лопаточно-плечевой дистрофией» . Ортопедия и травматология: хирургия и исследования . 106 (4): 701–707. дои : 10.1016/j.otsr.2020.03.002 . ПМИД 32430271 .
- ^ Теадом А., Родригес М., Роксбург Р., Балалла С., Хиггинс С., Бхаттачарджи Р., Джонс К., Кришнамурти Р., Фейгин В. (2014). «Распространенность мышечных дистрофий: систематический обзор литературы» . Нейроэпидемиология . 43 (3–4): 259–68. дои : 10.1159/000369343 . hdl : 10292/13206 . ПМИД 25532075 . S2CID 2426923 .
- ^ Мах Дж.К., Корнгут Л., Фиест К.М., Дайкман Дж., Дэй Л.Дж., Прингсхайм Т., Джетт Н. (январь 2016 г.). «Систематический обзор и метаанализ эпидемиологии мышечных дистрофий» . Канадский журнал неврологических наук . 43 (1): 163–77. дои : 10.1017/cjn.2015.311 . ПМИД 26786644 . S2CID 24936950 .
- ^ Перейти обратно: а б с д и ж г час я дж к Тавил Р., Ван дер Маарел С.М. (июль 2006 г.). «Лице-лопаточно-плечевая мышечная дистрофия» (PDF) . Мышцы и нервы . 34 (1): 1–15. дои : 10.1002/mus.20522 . ПМИД 16508966 . S2CID 43304086 .
- ^ Перейти обратно: а б с д Стэтленд Дж. М., Тавил Р. (декабрь 2016 г.). «Лице-лопаточно-плечевая мышечная дистрофия» . Континуум (Миннеаполис, Миннесота) . 22 (6, Заболевания мышц и нервно-мышечных соединений): 1916–1931. дои : 10.1212/CON.0000000000000399 . ПМЦ 5898965 . ПМИД 27922500 .
- ^ Перейти обратно: а б Крювейлье Ж (1852–1853). «Память об атрофическом мышечном параличе». Вестники Медицинской Академии . 18 : 490–502, 546–583.
- ^ Перейти обратно: а б с д Роджерс М.Т. (2004). «Лице-лопаточно-плечевая мышечная дистрофия: историческая справка и обзор литературы». В Упадхьяя М., Купер Д.Н. (ред.). Лице-лопаточно-плечевая мышечная дистрофия ЛЛПД: клиническая медицина и молекулярно-клеточная биология . Научные издательства БИОС. ISBN 1-85996-244-0 .
- ^ Перейти обратно: а б Ландузи Л., Дежерин Ж (1885). Ландузи Л., Лепин Р. (ред.). «Прогрессирующая атрофическая миопатия (миопатия без нейропатии, обычно начинающаяся в детстве на лице)» . Revue de Médecine (на французском языке). 5 . Феликс Алькан: 253–366 . Проверено 19 мая 2020 г.
- ^ Перейти обратно: а б с Коэн Дж., Дезимоун А., Лек М., Лек А. (октябрь 2020 г.). «Терапевтические подходы при лице-лопаточно-плечевой мышечной дистрофии» . Тенденции молекулярной медицины . 27 (2): 123–137. doi : 10.1016/j.molmed.2020.09.008 . ПМК 8048701 . ПМИД 33092966 .
- ^ Перейти обратно: а б Таплер Р., Барбьерато Л. и др. (сентябрь 1998 г.). «Идентичная мутация de novo в локусе D4F104S1 у монозиготных близнецов мужского пола, страдающих лице-лопаточно-плечевой мышечной дистрофией (ЛЛПД) с различным клиническим выражением» . Журнал медицинской генетики . 35 (9): 778–783. дои : 10.1136/jmg.35.9.778 . ПМЦ 1051435 . ПМИД 9733041 .
- ^ Тавил Р., Сторвик Д., Фисби Т.Э., Вайффенбах Б., Григгс Р.К. (февраль 1993 г.). «Чрезвычайная вариабельность экспрессии у монозиготных близнецов с мышечной дистрофией ФСГ». Неврология . 43 (2): 345–8. дои : 10.1212/wnl.43.2.345 . ПМИД 8094896 . S2CID 44422140 .
- ^ Пандия С., Эйхингер К. «Физическая терапия лице-лопаточно-плечевой мышечной дистрофии» (PDF) . Общество ФШД. Архивировано из оригинала (PDF) 14 апреля 2020 года . Проверено 14 апреля 2020 г.
- ^ Перейти обратно: а б с д и Падберг Г.В. (13 октября 1982 г.). Лице-лопаточно-плечевая болезнь (Диссертация). Лейденский университет.
- ^ Перейти обратно: а б с д и ж г час я дж к л Падберг Г.В. (2004). «Лице-лопаточно-плечевая мышечная дистрофия: опыт клинициста». В Упадхьяя М., Купер Д.Н. (ред.). Лице-лопаточно-плечевая мышечная дистрофия ЛЛПД: клиническая медицина и молекулярно-клеточная биология . Научные издательства БИОС. ISBN 1-85996-244-0 .
- ^ Перейти обратно: а б с Райкен Н.Х., ван дер Коой Э.Л., Хендрикс Дж.К., ван Ассельдонк Р.Дж., Падберг Г.В., Гертс А.К., ван Энгелен Б.Г. (декабрь 2014 г.). «Визуализация скелетных мышц при лице-лопаточно-плечевой мышечной дистрофии, характер и асимметрия поражения отдельных мышц» . Нервно-мышечные расстройства . 24 (12): 1087–96. дои : 10.1016/j.nmd.2014.05.012 . ПМИД 25176503 . S2CID 101093 .
- ^ Бергсма А, Cup EH, Янссен ММ, Гертс AC, де Гроот IJ (февраль 2017 г.). «Функция и активность верхних конечностей у людей с лице-лопаточно-плечевой мышечной дистрофией: интернет-опрос» . Инвалидность и реабилитация . 39 (3): 236–243. дои : 10.3109/09638288.2016.1140834 . ПМИД 26942834 . S2CID 4237308 .
- ^ Перейти обратно: а б с д и ж г час я дж к л м н тот Тавил Р., ван дер Маарел С.М., Тапскотт С.Дж. (10 июня 2014 г.). «Лице-лопаточно-плечевая дистрофия: путь к консенсусу по патофизиологии» . Скелетная мышца . 4 (1): 12. дои : 10.1186/2044-5040-4-12 . ПМК 4060068 . ПМИД 24940479 .
- ^ Цзя Ф.Ф., Дрю А.П., Николсон Г.А., Корбетт А., Кумар К.Р. (2 октября 2021 г.). «Лице-лопаточно-плечевая мышечная дистрофия типа 2: обновленная информация о клинических, генетических и молекулярных данных» . Нервно-мышечные расстройства . 31 (11): 1101–1112. дои : 10.1016/j.nmd.2021.09.010 . ПМИД 34711481 . S2CID 238246093 .
- ^ Перейти обратно: а б Упадхьяя М., Купер Д., ред. (март 2004 г.). Лице-лопаточно-плечевая мышечная дистрофия ЛЛПД: клиническая медицина и молекулярно-клеточная биология . Научные издательства БИОС. ISBN 0-203-48367-7 .
- ^ Перейти обратно: а б Голос пациента 29 июня 2020 года FDA провело внешнее совещание по разработке лекарств, ориентированных на пациентов, с участием около 400 пациентов с ЛЛПД, в результате которого был принят основополагающий документ под названием « » . В отчете отражено влияние ЛЛПД на повседневную жизнь людей с этим заболеванием, о котором говорят сами пациенты. Хотя ЛЛПД часто воспринимается как «легкое» заболевание, отчет показывает, что более 80% пациентов сообщают о «умеренном» или «сильном» ограничении повседневной деятельности.
- Общество F. «Общество FSHD публикует отчет «Голос пациента» о мышечной дистрофии FSH» . www.prweb.com . Проверено 31 января 2024 г.
- ^ Перейти обратно: а б Общество F. «Сообщество лицело-лопаточно-плечевой мышечной дистрофии обращается к FDA» . www.prweb.com . Проверено 31 января 2024 г.
- ^ Перейти обратно: а б Голос форума пациентов — Совещание по разработке лекарств, ориентированных на пациентов , 29 июня 2020 г. , получено 31 января 2024 г.
- ^ Перейти обратно: а б Оверман Д. (13 ноября 2020 г.). «Люди, живущие с ЛЛПД, рассказывают свои истории в новом отчете» . Управление реабилитацией . Проверено 31 января 2024 г.
- ^ Перейти обратно: а б Мул К., Берггрен К.Н., Силлс М.Ю., Маккалли А., ван Энгелен Б., Джонсон Н.Е., Стэтленд Дж.М. (26 февраля 2019 г.). «Влияние слабости орофациальных мышц на глотание и общение при ЛЛПД» . Неврология . 92 (9): е957–е963. дои : 10.1212/WNL.0000000000007013 . ПМК 6404471 . ПМИД 30804066 .
- ^ Вольгемут М., де Сварт Б.Дж., Кальф Дж.Г., Йоостен Ф.Б., Ван дер Влит А.М., Падберг Г.В. (27 июня 2006 г.). «Дисфагия при лице-лопаточно-плечевой мышечной дистрофии». Неврология . 66 (12): 1926–8. дои : 10.1212/01.wnl.0000219760.76441.f8 . ПМИД 16801662 . S2CID 7695047 .
- ^ Перейти обратно: а б «Гигант исследований ЛЛПД делится своими «сожалениями» » . Общество ФШД . Машина обратного пути. 30 сентября 2020 г. Архивировано из оригинала 24 октября 2020 г. Проверено 11 марта 2021 г.
Еще одним поразительным аспектом ЛЛПД является то, что мышечная слабость сильно варьируется от пациента к пациенту. Тем не менее, «существует весьма характерная картина мышечной слабости, иначе мы бы никогда не смогли распознать ЛЛПД как специфическое заболевание», — сказал Падберг. «Сильная дельтовидная мышца не возникает ни при каком другом состоянии, которое связано со слабостью стабилизаторов лопатки. Никакое другое заболевание мышц с поражением плечевого пояса не имеет такой закономерности». К сожалению, «объяснение нам не под силу, поскольку мы не знаем, как укладываются мышцы» на ранних стадиях беременности человека.
- ^ Перейти обратно: а б с Таска Г., Монфорте М., Ианнакконе Е., Лашена Ф., Оттавиани П., Леончини Е., Бочча С., Галлуцци Г., Пелличчони М., Маскулло М., Фрушанте Р., Меркури Е., Риччи Е. (2014). «Визуализация верхнего пояса при лице-лопаточно-плечевой мышечной дистрофии» . ПЛОС ОДИН . 9 (6): e100292. Бибкод : 2014PLoSO...9j0292T . дои : 10.1371/journal.pone.0100292 . ПМК 4059711 . ПМИД 24932477 .
- ^ Перейти обратно: а б с Геревини С, Скарлато М, Магги Л, Кава М, Кальендо Дж, Пасаниси Б, Фалини А, Превитали СК, Моранди Л (март 2016 г.). «Данные МРТ мышц при лице-лопаточно-плечевой мышечной дистрофии». Европейская радиология . 26 (3): 693–705. дои : 10.1007/s00330-015-3890-1 . ПМИД 26115655 . S2CID 24650482 .
- ^ Фо-Соловей А., Кульшреста Р., Эмери Н., Пандьян А., Уиллис Т., Филп Ф. (сентябрь 2021 г.). «Реабилитация верхних конечностей при фасцио-лопаточно-плечевой дистрофии (ЛЛПД): взгляд пациентов» . Архивы реабилитационных исследований и клинических переводов . 3 (4): 100157. doi : 10.1016/j.arrct.2021.100157 . ISSN 2590-1095 . ПМЦ 8683863 . ПМИД 34977539 .
- ^ Пандия С., Кинг В.М., Тавил Р. (1 января 2008 г.). «Лице-лопаточно-плечевая дистрофия» . Физиотерапия . 88 (1): 105–113. дои : 10.2522/ptj.20070104 . ПМИД 17986494 .
- ^ Лью В.К., ван дер Маарел С.М., Тавил Р. (2015). «Лице-лопаточно-плечевая дистрофия». Нервно-мышечные расстройства младенчества, детства и подросткового возраста . стр. 620–630. дои : 10.1016/B978-0-12-417044-5.00032-9 . ISBN 978-0-12-417044-5 .
- ^ Перейти обратно: а б с д и ж Гозелинк Р., Шреур В., ван Кернебек Ч.Р., Падберг Г.В., ван дер Маарель С.М., ван Энгелен Б., Эразмус К.Э., Тилен Т. (2019). «Офтальмологические данные при лице-лопаточно-плечевой дистрофии» . Мозговые коммуникации . 1 (1): fcz023. doi : 10.1093/braincomms/fcz023 . ПМЦ 7425335 . ПМИД 32954265 .
- ^ Падберг Г.В., Брауэр О.Ф., де Кайзер Р.Дж., Дейкман Г., Вейменга С., Гроте Дж.Дж., Франц Р.Р. (1995). «О значении заболеваний сосудов сетчатки и тугоухости при лице-лопаточно-плечевой мышечной дистрофии». Мышцы и нервы . 18 (С13): С73–С80. дои : 10.1002/mus.880181314 . hdl : 2066/20764 . S2CID 27523889 .
- ^ Линднер М., Хольц Ф.Г., Шарбель Исса П. (27 апреля 2016 г.). «Спонтанное разрешение сосудистых аномалий сетчатки и макулярного отека при лице-лопаточно-плечевой мышечной дистрофии». Клиническая и экспериментальная офтальмология . 44 (7): 627–628. дои : 10.1111/ceo.12735 . ISSN 1442-6404 . ПМИД 26933772 . S2CID 204996841 .
- ^ Тревизан КП, Пасторелло Э, Томеллери Г, Верчелли Л, Бруно С, Скаполан С, Сицилиано Г, Комаккио Ф (декабрь 2008 г.). «Лице-лопаточно-плечевая мышечная дистрофия: потеря слуха и другие атипичные особенности пациентов с большими делециями 4q35». Европейский журнал неврологии . 15 (12): 1353–8. дои : 10.1111/j.1468-1331.2008.02314.x . ПМИД 19049553 . S2CID 26276887 .
- ^ Перейти обратно: а б Эрен И, Абай Б, Гюнербююк С, Чакмак ОО, Шар С, Демирхан М (февраль 2020 г.). «Спондилодез при лице-лопаточно-плечевой дистрофии при гиперлордозе: описание случая» . Лекарство . 99 (8): e18787. дои : 10.1097/MD.0000000000018787 . ПМК 7034682 . ПМИД 32080072 .
- ^ Хумл Р.А., Перес Д.П. (2015). «ЛЛПД: наиболее распространенный тип мышечной дистрофии?». Мышечная дистрофия . стр. 9–19. дои : 10.1007/978-3-319-17362-7_3 . ISBN 978-3-319-17361-0 .
- ^ Вольгемут М., ван дер Коой Э.Л., ван Кестерен Р.Г., ван дер Маарель С.М., Падберг Г.В. (2004). «Вентиляционная поддержка при лице-лопаточно-плечевой мышечной дистрофии». Неврология . 63 (1): 176–8. CiteSeerX 10.1.1.543.2968 . дои : 10.1212/01.wnl.0000133126.86377.e8 . ПМИД 15249635 . S2CID 31335126 .
- ^ Даулинг Дж. Дж., Вейль К.С., Спенсер М.Дж. (ноябрь 2021 г.). «Молекулярные и клеточные основы генетически наследственных заболеваний скелетных мышц» . Обзоры природы. Молекулярно-клеточная биология . 22 (11): 713–732. дои : 10.1038/s41580-021-00389-z . ПМЦ 9686310 . ПМИД 34257452 . S2CID 235822532 .
- ^ Перейти обратно: а б с д и ж г Джаганнатан С. (1 мая 2022 г.). «Эволюция регуляции гена DUX4 и ее влияние на лице-лопаточно-плечевую мышечную дистрофию» . Biochimica et Biophysica Acta (BBA) - Молекулярные основы болезней . 1868 (5): 166367. doi : 10.1016/j.bbadis.2022.166367 . ПМК 9173005 . ПМИД 35158020 .
- ^ Перейти обратно: а б с д и ж г Леммерс Р.Дж., О'Ши С., Падберг Г.В., Лант П.В., ван дер Маарел С.М. (май 2012 г.). «Руководство по передовой практике генетической диагностики лице-лопаточно-плечевой мышечной дистрофии: семинар, 9 июня 2010 г., LUMC, Лейден, Нидерланды» . Нервно-мышечные расстройства . 22 (5): 463–470. дои : 10.1016/j.nmd.2011.09.004 . ПМИД 22177830 . S2CID 39898514 .
- ^ Название «D4Z4» происходит от устаревшей системы номенклатуры, используемой для сегментов ДНК неизвестного значения во время проекта генома человека : D для ДНК, 4 для хромосомы 4, Z указывает, что это повторяющаяся последовательность, а 4 — это серийный номер, присвоенный на основе о порядке подачи.
- Уайт Дж., Макэлпайн П., Антонаракис С., Канн Х., Эппиг Дж., Фрейзер К., Фрезал Дж., Ланцет Д., Намиас Дж., Пирсон П., Питерс Дж., Скотт А., Скотт Х., Сперр Н., Талбот С., Пови С. (октябрь 1997 г.) ). «НОМЕНКЛАТУРА» . Геномика . 45 (2): 468–471. дои : 10.1006/geno.1997.4979 . ПМИД 9344684 .
- Фасман К.Х., Летовский С.И., Коттингем Р.В., Кингсбери Д.Т. (1 января 1996 г.). «Усовершенствования базы данных генома человека GDB» . Исследования нуклеиновых кислот . 24 (1): 57–63. дои : 10.1093/нар/24.1.57 . ПМК 145602 . ПМИД 8594601 .
- ^ Перейти обратно: а б с д Невозможные вещи: зазеркалье с исследователями дистрофии ФСГ , Маргарет Уол, MDA , журнал Quest, том 14, № 2, март – апрель 2007 г.
- ^ Диксит М., Анссо Э., Тассин А. и др. (ноябрь 2007 г.). «DUX4, ген-кандидат лице-лопаточно-плечевой мышечной дистрофии, кодирует активатор транскрипции PITX1» . Учеб. Натл. акад. наук. США . 104 (46): 18157–62. Бибкод : 2007PNAS..10418157D . дои : 10.1073/pnas.0708659104 . ПМК 2084313 . ПМИД 17984056 .
- ^ Перейти обратно: а б с д и ж Шикрова Д., Теста А.М., Виллемсен И., ван ден Хеувел А., Тапскотт С.Дж., Даксингер Л., Балог Дж., ван дер Маарел С.М. (28 июня 2023 г.). «SMCHD1 и LRIF1 сходятся в ассоциированном с FSHD повторе D4Z4 и промоторе LRIF1, но демонстрируют разные способы действия» . Коммуникационная биология . 6 (1): 677. дои : 10.1038/s42003-023-05053-0 . ПМЦ 10307901 . ПМИД 37380887 .
- ^ Коппе Ф, Маттеотти С, Анссау Э, Соваж С, Леклерк И, Лерой А, Маркович А, Жербо С, Фиглевич Д, Дин Х, Белайев Б (2004). «Семейство генов DUX и ЛЛПД». В Упадхьяя М., Купер Д.Н. (ред.). Лице-лопаточно-плечевая мышечная дистрофия ЛЛПД: клиническая медицина и молекулярно-клеточная биология . Научные издательства БИОС. ISBN 1-85996-244-0 .
- ^ Росси М., Риччи Э., Колантони Л. и др. (2007). «Область лице-лопаточно-плечевой мышечной дистрофии на 4 четверти и гомологичный локус на 10 четверти развивались независимо под разным эволюционным давлением» . БМК Мед. Жене . 8 :8. дои : 10.1186/1471-2350-8-8 . ПМК 1821008 . ПМИД 17335567 .
- ^ Леммерс Р., ван дер Влит П.Дж., Блатник А., Балог Дж., Зидар Дж., Хендерсон Д., Гозелинк Р., Тапскотт С.Дж., Воерманс Н.К., Тавил Р., Падберг Г., ван Энгелен Б.Г., ван дер Маарел С.М. (12 января 2021 г.). «ЛМПД, связанный с хромосомой 10q, идентифицирует DUX4 как основной ген заболевания» . Журнал медицинской генетики . 59 (2): jmedgenet-2020-107041. doi : 10.1136/jmedgenet-2020-107041 . ПМЦ 8273184 . ПМИД 33436523 . S2CID 231589589 .
- ^ Перейти обратно: а б с Леммерс Р.Дж., ван дер Влит П.Дж., Балог Дж., Гоеман Дж.Дж., Ариндрарто В., Кром Ю.Д., Страашейм К.Р., Дебиперсад Р.Д., Озель Г., Соуден Дж., Снайдер Л., Мул К., Саккони С., ван Энгелен Б., Тапскотт С.Дж., Тавил Р. , ван дер Маарел С.М. (январь 2018 г.). «Глубокая характеристика распространенного варианта D4Z4 идентифицирует двуаллельную экспрессию DUX4 как модификатор пенетрантности заболевания при ЛЛПД2» . Европейский журнал генетики человека . 26 (1): 94–106. дои : 10.1038/s41431-017-0015-0 . ПМЦ 5838976 . ПМИД 29162933 .
- ^ Сидлаускайте Э, Ле Галль Л, Марио В, Дюмонсо Ж (28 июля 2020 г.). «Экспрессия DUX4 в мышцах ЛЛПД: внимание к регуляции его мРНК» . Журнал персонализированной медицины . 10 (3): 73. дои : 10.3390/jpm10030073 . ПМЦ 7564753 . ПМИД 32731450 .
- ^ Химеда CL, Джонс PL (31 августа 2019 г.). «Генетика и эпигенетика лице-лопаточно-плечевой мышечной дистрофии» . Ежегодный обзор геномики и генетики человека . 20 : 265–291. doi : 10.1146/annurev-genom-083118-014933 . ПМИД 31018108 . S2CID 131775712 .
- ^ Перейти обратно: а б Cabianca DS, Casa C, Bodega B и др. (11 мая 2012 г.). «Длинная нкРНК связывает изменение числа копий с эпигенетическим переключением полигребенчатости/триторакса при мышечной дистрофии ЛЛПД» . Клетка . 149 (4): 819–831. дои : 10.1016/j.cell.2012.03.035 . ПМЦ 3350859 . ПМИД 22541069 .
- ^ Перейти обратно: а б с д и ж Саккони С, Бриан-Сюло А, Грос М, Бодуэн С, Леммерс Р, Рондо С, Лага Н, Нигуманн П, Камбьери С, Пума А, Шапон Ф, Стойкович Т, Виал С, Бухур Ф, Као М, Пегораро Е, Петио П., Бехин А., Марк Б., Эймард Б., Эчанис-Лагуна А., Лафоре П., Сальвиати Л., Жанпьер М., Кристофари Г., ван дер Маарель С.М. (7 мая 2019 г.). «ЛЛПД1 и ЛЛПД2 образуют континуум заболевания» . Неврология . 92 (19): е2273–е2285. дои : 10.1212/WNL.0000000000007456 . ПМК 6537132 . ПМИД 30979860 .
- ^ Таплер Р., Берардинелли А., Барбьерато Л., Франц Р., Хьюитт Дж. Э., Ланци Г., Мараскио П., Тьеполо Л. (май 1996 г.). «Моносомия дистального сегмента 4q не вызывает лице-лопаточно-плечевой мышечной дистрофии» . Журнал медицинской генетики . 33 (5): 366–70. дои : 10.1136/jmg.33.5.366 . ПМК 1050603 . ПМИД 8733044 .
- ^ Тавил Р., Форрестер Дж., Григгс Р.К., Менделл Дж., Киссель Дж., Макдермотт М., Кинг В., Вайффенбах Б., Фиглевич Д. (июнь 1996 г.). «Доказательства ожидания и связи размера делеции с тяжестью лице-лопаточно-плечевой мышечной дистрофии. Группа FSH-DY». Анналы неврологии . 39 (6): 744–8. дои : 10.1002/ana.410390610 . ПМИД 8651646 . S2CID 84518968 .
- ^ Зернов Н., Скоблов М. (13 марта 2019 г.). «Генотип-фенотипические корреляции при ЛЛПД» . BMC Медицинская Геномика . 12 (Приложение 2): 43. doi : 10.1186/s12920-019-0488-5 . ПМК 6416831 . ПМИД 30871534 .
- ^ Саккони С., Сальвиати Л., Деснуэль К. (апрель 2015 г.). «Лице-лопаточно-плечевая мышечная дистрофия» . Biochimica et Biophysica Acta (BBA) - Молекулярные основы болезней . 1852 (4): 607–14. дои : 10.1016/j.bbadis.2014.05.021 . ПМИД 24882751 .
- ^ Перейти обратно: а б с Леммерс Р.Дж., Тавил Р., Петек Л.М. и др. (декабрь 2012 г.). «Дигенное наследование мутации SMCHD1 и FSHD-пермиссивного аллеля D4Z4 вызывает лице-лопаточно-плечевую мышечную дистрофию 2-го типа» . Природная генетика . 44 (12): 1370–1374. дои : 10.1038/ng.2454 . ПМК 3671095 . ПМИД 23143600 .
- ^ ван ден Бугаард М.Л., Леммерс Р., Балог Дж., Вольгемут М., Ауранен М., Мицухаши С., ван дер Влит П.Дж., Страшейм К.Р., ван ден Аккер Р., Крик М., Лоуренс-Бик М., Раз В., ван Остайен-Тен Дам М.М. , Ханссон К., ван дер Коой Э.Л., Киуру-Энари С., Удд Б., ван Тол М., Нишино И., Тавил Р., Тапскотт С.Дж., ван Энгелен Б., ван дер Маарел С.М. (5 мая 2016 г.). «Мутации в DNMT3B изменяют эпигенетическую репрессию повтора D4Z4 и пенетрантность лице-лопаточно-плечевой дистрофии» . Американский журнал генетики человека . 98 (5): 1020–1029. дои : 10.1016/j.ajhg.2016.03.013 . ПМЦ 4863565 . ПМИД 27153398 .
- ^ Перейти обратно: а б Джонсон Н.Е., Стэтленд Дж.М. (7 мая 2019 г.). «FSHD1 или FSHD2: вот в чем вопрос: ответ: это все просто FSHD». Неврология . 92 (19): 881–882. дои : 10.1212/WNL.0000000000007446 . ПМИД 30979855 . S2CID 111390628 .
- ^ Перейти обратно: а б с Хаманака К, Ксикрова Д, Мицухаси С, Масуда Х, Секигути Ю, Сугияма А, Сибуя К, Леммерс Р.Дж., Гуссенс Р, Огава М, Нагао К, Обусе С, Ногучи С, Хаяси Ю.К, Кувабара С, Балог Дж, Нишино И , автор Der Maarel SM (28 мая 2020 г.). «Гомозиготный нонсенс-вариант, связанный с лице-лопаточно-плечевой мышечной дистрофией» . Неврология . 94 (23): е2441–е2447. дои : 10.1212/WNL.0000000000009617 . ПМЦ 7455367 . ПМИД 32467133 . S2CID 218982743 .
- ^ Шеффер А.А. (октябрь 2013 г.). «Дигенная наследственность в медицинской генетике» . Журнал медицинской генетики . 50 (10): 641–52. doi : 10.1136/jmedgenet-2013-101713 . ПМК 3778050 . ПМИД 23785127 .
- ^ Перейти обратно: а б Капуто V, Мегалицци Д, Фабрицио С, Термине А, Колантони Л, Кальтаджироне С, Джардина Е, Касчелла Р, Страфелла С (29 августа 2022 г.). «Обновленная информация о молекулярных аспектах и методах, лежащих в основе сложной архитектуры ЛЛПД» . Клетки . 11 (17): 2687. doi : 10.3390/cells11172687 . ПМЦ 9454908 . ПМИД 36078093 .
- ^ Перейти обратно: а б с Саккони С., Леммерс Р.Дж., Балог Дж. и др. (3 октября 2013 г.). «Ген FSHD2 SMCHD1 является модификатором тяжести заболевания в семьях, затронутых FSHD1» . Американский журнал генетики человека . 93 (4): 744–751. дои : 10.1016/j.ajhg.2013.08.004 . ПМЦ 3791262 . ПМИД 24075187 .
- ^ Перейти обратно: а б Лим К., Нгуен К., Йокота Т. (22 января 2020 г.). «Передача сигналов DUX4 в патогенезе лице-лопаточно-плечевой мышечной дистрофии» . Международный журнал молекулярных наук . 21 (3): 729. doi : 10.3390/ijms21030729 . ПМК 7037115 . ПМИД 31979100 .
- ^ Перейти обратно: а б Боснаковски Д., Шамс А.С., Юань С., да Силва М.Т., Энер Э.Т., Бауманн К.В., Линдси А.Дж., Верма М., Асакура А., Лоу Д.А., Киба М. (6 апреля 2020 г.). «Транскрипционные и цитопатологические признаки ЛЛПД у мышей с хронической экспрессией DUX4» . Журнал клинических исследований . 130 (5): 2465–2477. дои : 10.1172/JCI133303 . ПМК 7190912 . ПМИД 32250341 .
- ^ Моччаро Э., Рунфола В., Гецци П., Паннезе М., Габеллини Д. (26 ноября 2021 г.). «Роль DUX4 в нормальной физиологии и мышечной дистрофии ЛЛПД» . Клетки . 10 (12): 3322. doi : 10.3390/cells10123322 . ПМЦ 8699294 . ПМИД 34943834 .
- ^ Перейти обратно: а б Шецль Т., Кайзер Л., Дейнер Х.П. (12 марта 2021 г.). «Лице-лопаточно-плечевая мышечная дистрофия: генетика, активация генов и последующая передача сигналов в отношении последних терапевтических подходов: обновленная информация» . Сиротский журнал редких заболеваний . 16 (1): 129. дои : 10.1186/s13023-021-01760-1 . ПМЦ 7953708 . ПМИД 33712050 . S2CID 232202360 .
- ^ Лек А., Чжан Ю., Вудман К.Г., Хуанг С., Дезимоун А.М., Коэн Дж., Хо В., Коннер Дж., Мид Л., Кодани А., Пакула А., Санджана Н., Кинг О.Д., Джонс П.Л., Вагнер К.Р., Лек М., Кункель Л.М. (25 марта 2020 г.). «Применение полногеномного скрининга CRISPR-Cas9 для открытия терапевтических средств при лице-лопаточно-плечевой мышечной дистрофии» . Наука трансляционной медицины . 12 (536): eaay0271. doi : 10.1126/scitranslmed.aay0271 . ПМК 7304480 . ПМИД 32213627 .
- ^ Марио В., Жубер Р., Ле Галл Л., Сидлаускайте Е., Урд К., Дадди В., Войт Т., Бенце М., Дюмонсо Ж. (22 октября 2021 г.). «RIPK3-опосредованная гибель клеток участвует в DUX4-опосредованной токсичности при лице-лопаточно-плечевой дистрофии» . Журнал кахексии, саркопении и мышц . 12 (6): 2079–2090. дои : 10.1002/jcsm.12813 . ПМЦ 8718031 . ПМИД 34687171 . S2CID 239471655 .
- ^ Ван Л.Х., Фридман С.Д., Шоу Д., Снайдер Л., Вонг С.Дж., Будеч С.Б., Полячик С.Л., Гоув Н.Е., Льюис Л.М., Кэмпбелл А.Е., Леммерс Р., Маарел С.М., Тапскотт С.Дж., Тавил Р.Н. (01.02.2019). «Биопсия мышц, полученная с помощью МРТ, коррелирует с патологией и экспрессией гена-мишени DUX4 при ЛЛПД» . Молекулярная генетика человека . 28 (3): 476–486. дои : 10.1093/hmg/ddy364 . ПМК 6337697 . ПМИД 30312408 .
- ^ Герарди Р., Амато А.А., Лидов Х.Г., Джиролами УД (ноябрь 2018 г.). «Патология скелетных мышц». В Грей Ф, Дуйкертс С, Джиролами Вуд (ред.). Руководство Эскуроля и Пуарье по базовой невропатологии (Шестое изд.). Нью-Йорк, штат Нью-Йорк: Издательство Оксфордского университета. дои : 10.1093/med/9780190675011.001.0001 . ISBN 978-0-19-067501-1 .
- ^ ван дер Маарель С.М., Миллер Д.Г., Тавил Р., Филиппова Г.Н., Тапскотт С.Дж. (октябрь 2012 г.). «Лице-лопаточно-плечевая мышечная дистрофия: последствия релаксации хроматина» . Современное мнение в неврологии . 25 (5): 614–20. дои : 10.1097/WCO.0b013e328357f22d . ПМЦ 3653067 . ПМИД 22892954 .
- ^ Перейти обратно: а б с Майр Д., Хьюгенс-Пензель М., Кресс В., Рот С., Ферберт А. (2017). «Поражение мышц ног при лице-лопаточно-плечевой мышечной дистрофии: сравнение типов лице-лопаточно-плечевой мышечной дистрофии 1 и 2». Европейская неврология . 77 (1–2): 32–39. дои : 10.1159/000452763 . ПМИД 27855411 . S2CID 25005883 .
- ^ Перейти обратно: а б с Олсен Д.Б., Гидеон П., Джеппесен Т.Д., Виссинг Дж. (ноябрь 2006 г.). «Поражение мышц ног при лице-лопаточно-плечевой мышечной дистрофии по данным МРТ». Журнал неврологии . 253 (11): 1437–41. дои : 10.1007/s00415-006-0230-z . ПМИД 16773269 . S2CID 19421344 .
- ^ Саккони С., Сальвиати Л., Бурже И., Фигарелла Д., Переон Ю., Леммерс Р., ван дер Маарель С., Деснуэль К. (24 октября 2006 г.). «Проблемы диагностики лице-лопаточно-плечевой мышечной дистрофии». Неврология . 67 (8): 1464–6. дои : 10.1212/01.wnl.0000240071.62540.6f . hdl : 11577/1565214 . ПМИД 17060574 . S2CID 25693278 .
- ^ Страфелла С, Капуто В, Галота Р.М., Камполи Г, Бакс С, Колантони Л, Миноцци Г, Орсини С, Политано Л, Таска Г, Новелли Г, Риччи Е, Джардина Е, Касчелла Р (1 декабря 2019 г.). «Вариабельность гена SMCHD1 у пациентов с ЛЛПД: свидетельства новых мутаций» . Молекулярная генетика человека . 28 (23): 3912–3920. дои : 10.1093/hmg/ddz239 . ПМК 6969370 . ПМИД 31600781 .
- ^ Зампатти С., Колантони Л., Страфелла С., Галота Р.М., Капуто В., Камполи Г., Пальяроли Дж., Карбони С., Мела Дж., Пекони С., Гамбарделла С., Каселла Р., Джардина Э. (май 2019 г.). «Молекулярная диагностика лице-лопаточно-плечевой мышечной дистрофии (ЛЛПД): от традиционных технологий до эпохи NGS». Нейрогенетика . 20 (2): 57–64. дои : 10.1007/s10048-019-00575-4 . ПМИД 30911870 . S2CID 85495566 .
- ^ Киносита Дж. (11 марта 2020 г.). «Генетическое тестирование ЛЛПД — новый рубеж» . Общество ФШД . Архивировано из оригинала 8 апреля 2020 года . Проверено 8 апреля 2020 г.
- ^ Васале Дж , Бояр Ф , Джоксон М , Сулцова В , Чан П , Лиакат К , Хоффман С , Месерви М , Чанг И , Цао Д , Хенсли К , Лю Ю , Оуэн Р , Браастад С , Сан В , Вальрафен П , Комацу Дж , Ван Дж.К., Бенсимон А., Ангиано А., Яремко М., Ван З., Батиш С., Стром С., Хиггинс Дж. (декабрь 2015 г.). «Молекулярное расчесывание по сравнению с Саузерн-блоттингом для измерения сокращений D4Z4 при ЛЛПД». Нервно-мышечные расстройства . 25 (12): 945–51. дои : 10.1016/j.nmd.2015.08.008 . ПМИД 26420234 . S2CID 6871094 .
- ^ Перейти обратно: а б с д Гулд Т., Джонс Т.И., Джонс П.Л. (13 августа 2021 г.). «Точный эпигенетический анализ с использованием целевого бисульфитного геномного секвенирования позволяет отличить FSHD1, FSHD2 и здоровых субъектов» . Диагностика (Базель, Швейцария) . 11 (8): 1469. doi : 10.3390/diagnostics11081469 . ПМЦ 8393475 . ПМИД 34441403 .
- ^ Перейти обратно: а б ван Дойтеком Дж.К., Вейменга С., ван Тьенховен Е.А. и др. (декабрь 1993 г.). «Перестройки ДНК, связанные с FSHD, происходят из-за делеций цельных копий тандемно повторяющейся единицы размером 3,2 т.п.н.». Молекулярная генетика человека . 2 (12): 2037–2042. дои : 10.1093/hmg/2.12.2037 . ПМИД 8111371 .
- ^ Перейти обратно: а б Вейменга С., Хьюитт Дж.Э., Сандкуйл Л.А. и др. (сентябрь 1992 г.). «Перестройки ДНК хромосомы 4q, связанные с лице-лопаточно-плечевой мышечной дистрофией». Природная генетика . 2 (1): 26–30. дои : 10.1038/ng0992-26 . ПМИД 1363881 . S2CID 21940164 .
- ^ Франц Р.Р., Сандкуйл Л.А., ван дер Маарель С.М., Падберг Г.В. (2004). «Картирование гена ЛЛПД и обнаружение патогномоничной делеции». В Упадхьяя М., Купер Д.Н. (ред.). Лице-лопаточно-плечевая мышечная дистрофия ЛЛПД: клиническая медицина и молекулярно-клеточная биология . Научные издательства БИОС. ISBN 1-85996-244-0 .
- ^ Леммерс Р.Дж., Влит П.Дж., Гранадо Д.С., Стоп Н., Буерманс Х., Шендель Р., Шиммель Дж., Виссер М., Костер Р., Жанпьер М., Лафоре П., Упадхьяя М., Энгелен Б., Саккони С., Тавил Р., Верманс Н.К., Роджерс М. , ван дер Маарель С.М. (24 сентября 2021 г.). «Картирование точек разрыва с высоким разрешением проксимально расширенных делеций D4Z4 при FSHD1 обнаруживает доказательства эффекта основателя» . Молекулярная генетика человека . 31 (5): 748–760. дои : 10.1093/hmg/ddab250 . ПМЦ 8895739 . ПМИД 34559225 .
- ^ Гайяр MC, Рош С, Дион С, Тасмаджян А, Буже Г, Салорт-Кампана Е, Вован С, Ше С, Бруксо Н, Морере Дж, Пуппо Ф, Бартоли М, Леви Н, Бернар Р, Аттариан С, Нгуен К, Магдинье Ф (19 августа 2014 г.). «Дифференциальное метилирование ДНК повтора D4Z4 у пациентов с ЛЛПД и бессимптомными носителями» (PDF) . Неврология . 83 (8): 733–42. дои : 10.1212/WNL.0000000000000708 . ПМИД 25031281 . S2CID 10002229 .
- ^ Информационный бюллетень FSHD. Архивировано 6 марта 2006 г. в Wayback Machine , MDA , 01.11.2001.
- ^ Перейти обратно: а б с д и ж Ваттджес М.П., Клей Р.А., Фишер Д. (октябрь 2010 г.). «Нейромышечная визуализация при наследственных заболеваниях мышц» . Европейская радиология . 20 (10): 2447–60. дои : 10.1007/s00330-010-1799-2 . ПМК 2940021 . ПМИД 20422195 .
- ^ Перейти обратно: а б Престон М.К., Тавил Р., Ван Л.Х., Адам М.П., Ардингер Х.Х., Пагон Р.А., Уоллес С.Е., Бин Л., Мирзаа Г., Амемия А. (1993). «Лице-лопаточно-плечевая мышечная дистрофия». ПМИД 20301616 .
{{cite journal}}: Для цитирования журнала требуется|journal=( помощь ) - ^ Перейти обратно: а б с д и ж г час Тавил Р., Киссель Дж.Т., Хитволе С., Пандия С., Гронсет Г., Бенатар М. (28 июля 2015 г.). «Краткий обзор научно обоснованного руководства: Оценка, диагностика и лечение лице-лопаточно-плечевой мышечной дистрофии: отчет подкомитета по разработке, распространению и внедрению рекомендаций Американской академии неврологии и группы по рассмотрению практических вопросов Американской ассоциации нейромышечной и электродиагностической медицины». " . Неврология . 85 (4): 357–64. дои : 10.1212/WNL.0000000000001783 . ПМК 4520817 . ПМИД 26215877 .
- ^ Перейти обратно: а б с Тавил Р., ван дер Маарел С., Падберг Г.В., ван Энгелен Б.Г. (июль 2010 г.). «171-й международный семинар ENMC: Стандарты лечения и лечения лице-лопаточно-плечевой мышечной дистрофии». Нервно-мышечные расстройства . 20 (7): 471–5. дои : 10.1016/j.nmd.2010.04.007 . ПМИД 20554202 . S2CID 18448196 .
- ^ Перейти обратно: а б Фут Н, Блейенберг Г, Хендрикс Дж, де Гроот И, Падберг Г, ван Энгелен Б, Гертс А (18 ноября 2014 г.). «И аэробные упражнения, и когнитивно-поведенческая терапия уменьшают хроническую усталость при ЛЛПД: РКИ». Неврология . 83 (21): 1914–22. дои : 10.1212/WNL.0000000000001008 . ПМИД 25339206 . S2CID 25382403 .
- ^ Перейти обратно: а б Янссен Б., Воет Н., Гертс А., ван Энгелен Б., Хеершап А. (3 мая 2016 г.). «Количественная МРТ выявляет замедленную жировую инфильтрацию в мышцах у активных пациентов с ЛЛПД». Неврология . 86 (18): 1700–7. дои : 10.1212/WNL.0000000000002640 . ПМИД 27037227 . S2CID 11617226 .
- ^ Voet NB, ван дер Коой Э.Л., ван Энгелен Б.Г., Гертс АС (6 декабря 2019 г.). «Силовые тренировки и аэробные упражнения при заболеваниях мышц» . Кокрановская база данных систематических обзоров . 2019 (12): CD003907. дои : 10.1002/14651858.CD003907.pub5 . ISSN 1469-493X . ПМК 6953420 . ПМИД 31808555 .
- ^ Перейти обратно: а б Тавил Р., Мах Дж.К., Бейкер С., Вагнер К.Р., Райан М.М., Sydney Workshop P (июль 2016 г.). «Аспекты клинической практики лице-лопаточно-плечевой мышечной дистрофии Сидней, Австралия, 21 сентября 2015 г.» . Нервно-мышечные расстройства . 26 (7): 462–71. дои : 10.1016/j.nmd.2016.03.007 . ПМИД 27185458 .
- ^ «Информация для пациентов и семей - Центр Ричарда Филдса по дистрофии ФСГ (ЛСГД) и нервно-мышечных исследований - Медицинский центр Университета Рочестера» . www.urmc.rochester.edu . Архивировано из оригинала 14 ноября 2019 года . Проверено 14 апреля 2020 г.
- ^ Априле И, Бордьери С, Джиларди А, Лайнери Милаццо М, Руссо Г, Де Сантис Ф, Фрушанте Р, Ианнакконе Е, Эрра С, Риччи Е, Падуя Л (апрель 2013 г.). «Нарушение равновесия и ходьба при лице-лопаточно-плечевой дистрофии: пилотное исследование влияния индивидуальных ортезов нижних конечностей». Европейский журнал физической и реабилитационной медицины . 49 (2): 169–78. ПМИД 23138679 .
- ^ Перейти обратно: а б с д и Оррелл Р.В., Коупленд С., Роуз М.Р. (20 января 2010 г.). «Фиксация лопатки при мышечной дистрофии» . Кокрейновская база данных систематических обзоров . 2010 (1): CD003278. дои : 10.1002/14651858.CD003278.pub2 . ПМЦ 7144827 . ПМИД 20091543 .
- ^ Демирхан М, Уйсал О, Аталар А.С., Кылыкоглу О, Сердароглу П (31 марта 2009 г.). «Лопато-грудной артродез при лице-лопаточно-плечевой дистрофии с использованием многофиламентного кабеля» . Клиническая ортопедия и связанные с ней исследования . 467 (8): 2090–2097. дои : 10.1007/s11999-009-0815-9 . ПМК 2706357 . ПМИД 19333668 .
- ^ ДеФранко М.Дж., Нхо С., Ромео А.А. (апрель 2010 г.). «Скапулоторакальный спондилодез». Журнал Американской академии хирургов-ортопедов . 18 (4): 236–42. дои : 10.5435/00124635-201004000-00006 . ПМИД 20357232 . S2CID 27456684 .
- ^ Хеллер К.Д., Прешер А., Форст Дж., Штадтмюллер А., Форст Р. (1996). «Анатомо-экспериментальное исследование шнурковой фиксации крылатой лопатки при мышечной дистрофии». Хирургическая и радиологическая анатомия . 18 (2): 75–9. дои : 10.1007/BF01795222 . ПМИД 8782311 . S2CID 20162712 .
- ^ Абрамс Дж.С., Белл Р.Х., Токиш Дж.М. (2018). РАСШИРЕННАЯ РЕКОНСТРУКЦИЯ ПЛЕЧА . АМЕРСКАЯ АКАДЕМИЯ ОРТОПЕДИИ. ISBN 978-1-9751-2347-5 .
- ^ Перейти обратно: а б Упадхьяя М., Купер Д.Н. (2004). «Введение и обзор ЛЛПД». В Упадхьяя М., Купер Д.Н. (ред.). Лице-лопаточно-плечевая мышечная дистрофия ЛЛПД: клиническая медицина и молекулярно-клеточная биология . Научные издательства БИОС. ISBN 1-85996-244-0 .
- ^ Сансоне В., Бойнтон Дж., Паленски С. (июнь 1997 г.). «Использование золотых гирь для коррекции лагофтальма при нервно-мышечных заболеваниях». Неврология . 48 (6): 1500–3. дои : 10.1212/wnl.48.6.1500 . hdl : 2434/210652 . ПМИД 9191754 . S2CID 16251273 .
- ^ Мацумото М., Онода С., Уэхара Х., Миура Ю., Катаяма Ю., Кимата Ю. (сентябрь 2016 г.). «Коррекция нижней губы хрящевым трансплантатом и резекцией губы у больных лице-лопаточно-плечевой мышечной дистрофией». Журнал черепно-лицевой хирургии . 27 (6): 1427–9. дои : 10.1097/SCS.0000000000002720 . ПМИД 27300465 . S2CID 16343571 .
- ^ Расинг Н.Б., ван де Геест-Буит В.А., Чан О.Ю., Мул К., Лансер А., ван Энгелен Б.Г., Эразмус К.Э., Фишер А.Х., Ингельс К.Дж., Пост Б, Симанн И., Гротуис Дж.Т., Фёрманс NC (21 марта 2024 г.). «Подходы к лечению измененного выражения лица: систематический обзор лице-лопаточно-плечевой мышечной дистрофии и других неврологических заболеваний» . Журнал нервно-мышечных заболеваний . 11 (3): 535–565. дои : 10.3233/JND-230213 . ПМЦ 11091602 . ПМИД 38517799 .
- ^ Кришнамурти С., Ибрагим М. (январь 2019 г.). «Перенос сухожилий при отвисании стопы» . Индийский журнал пластической хирургии . 52 (1): 100–108. дои : 10.1055/s-0039-1688105 . ПМК 6664842 . ПМИД 31456618 .
- ^ Чиодо С., Блюман Э.М. (21 октября 2011 г.). Трансплантация сухожилий стопы и голеностопного сустава . Сондерс. п. 421. ИСБН 978-1-4557-0924-3 . Проверено 1 января 2020 г.
- ^ Лунт П., Упадхьяя М., Кох М.С. (2004). «Генотип-фенотипические отношения при ЛЛПД». В Упадхьяя М., Купер Д.Н. (ред.). Лице-лопаточно-плечевая мышечная дистрофия ЛЛПД: клиническая медицина и молекулярно-клеточная биология . БИОС Сайентифик Паблишерс Лимитед. п. 157.
- ^ Винсентен СК, Мул К., Шредер Т.Х., Фёрманс НК, Хорлингс К.Г., ван Энгелен Б.Г. (июль 2021 г.). «Изучение влияния курения и употребления алкоголя на тяжесть клинической картины у пациентов с лице-лопаточно-плечевой мышечной дистрофией» . Нервно-мышечные расстройства . 31 (9): 824–828. дои : 10.1016/j.nmd.2021.07.005 . hdl : 2066/239258 . ISSN 0960-8966 . ПМИД 34407911 .
- ^ Перейти обратно: а б с Мэсси Дж. М., Гейбл К. Л. (1 февраля 2022 г.). «Нервно-мышечные расстройства и беременность». Континуум (Миннеаполис, Миннесота) . 28 (1): 55–71. дои : 10.1212/CON.0000000000001069 . ПМИД 35133311 . S2CID 246651681 .
- ^ Перейти обратно: а б с Динен Дж.К., Арнц Х., ван дер Маарель С.М., Падберг Г.В., Вершуурен Дж.Дж., Баккер Э., Вайнрайх СС, Вербек А.Л., ван Энгелен Б.Г. (2014). «Популяционная заболеваемость и распространенность лице-лопаточно-плечевой дистрофии» . Неврология . 83 (12): 1056–9. дои : 10.1212/WNL.0000000000000797 . ПМК 4166358 . ПМИД 25122204 .
- ^ Динен Дж.К., Хорлингс К.Г., Вершуурен Дж.Дж., Вербек А.Л., ван Энгелен Б.Г. (2015). «Эпидемиология нервно-мышечных расстройств: комплексный обзор литературы» . Журнал нервно-мышечных заболеваний . 2 (1): 73–85. дои : 10.3233/JND-140045 . ПМИД 28198707 .
- ^ Теверони Э, Пеллегрино М, Саккони С, Каландра П, Кашино И, Фариоли-Веккиоли С, Пума А, Гарибальди М, Морозетти Р, Таска Г, Риччи Е, Тревизан С.П., Галлуцци Г, Понтекорви А, Крещенци М, Дейдда Г, Моретти Ф (3 апреля 2017 г.). «Эстрогены усиливают дифференцировку миобластов при лице-лопаточно-плечевой мышечной дистрофии, противодействуя активности DUX4» . Журнал клинических исследований . 127 (4): 1531–1545. дои : 10.1172/JCI89401 . ПМЦ 5373881 . ПМИД 28263188 .
- ^ Мул К., Хорлингс С., Фёрманс Н.К., Шредер Т., ван Энгелен Б. (июнь 2018 г.). «Воздействие эндогенного эстрогена в течение жизни и тяжесть заболевания у пациенток с лице-лопаточно-плечевой мышечной дистрофией» . Нервно-мышечные расстройства . 28 (6): 508–511. дои : 10.1016/j.nmd.2018.02.012 . hdl : 2066/194350 . ПМИД 29655530 .
- ^ Дюшен ГБ (1868). «Псевдогипертрофический мышечный паралич, или миосклеротический паралич» . Арх. Ген. Мед. (на французском языке). 11 . Национальная библиотека Франции: 5–25, 179–209, 305–321, 421–443, 552–588 . Проверено 18 мая 2020 г.
- ^ Перейти обратно: а б Синдром Ландузи-Дежерина , whonamedit.com, дата обращения 11 марта 2007 г.
- ^ Ландузи, Дежерин (1884). «Прогрессирующая атрофическая миопатия (наследственная миопатия, начинающаяся в детском возрасте на лице, без поражения нервной системы)». Известия Академии наук . 98 : 53–55.
- ^ Ландузи, Дежерин (1886). «Вклад в изучение прогрессирующей атрофической миопатии (прогрессирующая атрофическая миопатия лопаточно-плечевого типа)». Отчеты заседаний Биологического общества . 38 : 478–481.
- ^ Тайлер Ф., Стивенс Ф.Е. (апрель 1950 г.). «Исследования заболеваний мышц. II Клинические проявления и наследование лице-лопаточно-плечевой дистрофии в большой семье». Анналы внутренней медицины . 32 (4): 640–660. дои : 10.7326/0003-4819-32-4-640 . ПМИД 15411118 .
- ^ Кениг М., Хоффман Э.П., Бертельсон С.Дж., Монако А.П., Финер С., Кункель Л.М. (31 июля 1987 г.). «Полное клонирование кДНК мышечной дистрофии Дюшенна (МДД) и предварительная геномная организация гена МДД у нормальных и больных людей» . Клетка . 50 (3): 509–517. дои : 10.1016/0092-8674(87)90504-6 . ПМИД 3607877 . S2CID 35668717 .
- ^ Вейменга С., Падберг Г.В., Мёрер П. и др. (апрель 1991 г.). «Картирование гена лице-лопаточно-плечевой мышечной дистрофии с хромосомой 4q35-qter посредством анализа многоточечного сцепления и гибридизации in situ». Геномика . 9 (4): 570–575. дои : 10.1016/0888-7543(91)90348-I . ПМИД 2037288 .
- ^ Гилберт Дж.Р., Стажич Дж.М., Уолл С. и др. (август 1993 г.). «Доказательства гетерогенности лице-лопаточно-плечевой мышечной дистрофии (ЛЛПД)» . Американский журнал генетики человека . 53 (2): 401–408. ПМК 1682358 . ПМИД 8328457 .
- ^ Перейти обратно: а б Винокур С.Т., Бенгтссон Ю., Феддерсен Дж. и др. (май 1994 г.). «Перестройка ДНК, связанная с лице-лопаточно-плечевой мышечной дистрофией, включает в себя повторяющийся элемент, связанный с гетерохроматином: значение роли структуры хроматина в патогенезе заболевания». Хромосомные исследования . 2 (3): 225–234. дои : 10.1007/bf01553323 . ПМИД 8069466 . S2CID 6933736 .
- ^ Хьюитт Дж.Э., Лайл Р., Кларк Л.Н. и др. (август 1994 г.). «Анализ локуса тандемных повторов D4Z4, связанного с лице-лопаточно-плечевой мышечной дистрофией». Молекулярная генетика человека . 3 (8): 1287–1295. дои : 10.1093/hmg/3.8.1287 . ПМИД 7987304 .
- ^ Гилберт Дж.Р., Спир М.К., Стажич Дж. и др. (октябрь 1995 г.). «Картирование исключения хромосомных областей, которые перекрестно гибридизуются с маркерами, связанными с FSHD1A, в FSHD1B» . Журнал медицинской генетики . 32 (10): 770–773. дои : 10.1136/jmg.32.10.770 . ПМЦ 1051697 . ПМИД 8558552 .
- ^ ван Дойтеком Дж.К., Леммерс Р.Дж., Гревал П.К. и др. (май 1996 г.). «Идентификация первого гена (FRG1) из области FSHD на хромосоме 4q35 человека» . Молекулярная генетика человека . 5 (5): 581–590. дои : 10.1093/hmg/5.5.581 . ПМИД 8733123 .
- ^ Перейти обратно: а б Ковальёв В., Маркович А., Ансо Э. и др. (август 2007 г.). «Ген DUX4 в локусе FSHD1A кодирует проапоптотический белок» . Нервно-мышечные расстройства . 17 (8): 611–623. дои : 10.1016/j.nmd.2007.04.002 . ПМИД 17588759 . S2CID 25926418 .
- ^ Габриэльс Дж., Беккерс М.К., Дин Х. и др. (5 августа 1999 г.). «Нуклеотидная последовательность частично удаленного локуса D4Z4 у пациента с ЛЛПД идентифицирует предполагаемый ген внутри каждого элемента размером 3,3 т.п.н.». Джин . 236 (1): 25–32. дои : 10.1016/S0378-1119(99)00267-X . ПМИД 10433963 .
- ^ Цянь Ф., Сунь Б., Хопкинс Н.Э. и др. (ноябрь 2001 г.). «Метилирование субтеломерного повтора, связанного с синдромом ЛЛПД, в нормальных клеточных культурах и тканях ЛЛПД». Молекулярная генетика и обмен веществ . 74 (3): 322–331. дои : 10.1006/mgme.2001.3219 . ПМИД 11708861 .
- ^ Леммерс Р.Дж., де Киевит П., Сандкуйл Л. и др. (октябрь 2002 г.). «Лице-лопаточно-плечевая мышечная дистрофия однозначно связана с одним из двух вариантов субтеломеры 4q». Природная генетика . 32 (2): 235–236. дои : 10.1038/ng999 . ПМИД 12355084 . S2CID 28107557 .
- ^ Габеллини Д., Грин М.Р., Туплер Р. (9 августа 2002 г.). «Несоответствующая активация генов при ЛЛПД: репрессорный комплекс связывает хромосомный повтор, удаленный в дистрофической мышце» . Клетка . 110 (3): 339–348. дои : 10.1016/S0092-8674(02)00826-7 . hdl : 11380/459475 . ПМИД 12176321 . S2CID 16396883 .
- ^ ван Овервельд П.Г., Леммерс Р.Дж., Сандкуйл Л.А. и др. (декабрь 2003 г.). «Гипометилирование D4Z4 при 4q-связанной и несвязанной 4q-связанной лице-лопаточно-плечевой мышечной дистрофии». Природная генетика . 35 (4): 315–317. дои : 10.1038/ng1262 . ПМИД 14634647 . S2CID 28696708 .
- ^ Леммерс Р.Дж., Вольгемут М., Франц Р.Р., Падберг Г.В., Морава Э., ван дер Маарел С.М. (декабрь 2004 г.). «Сокращения D4Z4 на субтеломерах 4qB не вызывают лице-лопаточно-плечевой мышечной дистрофии» . Американский журнал генетики человека . 75 (6): 1124–1130. дои : 10.1086/426035 . ПМЦ 1182148 . ПМИД 15467981 .
- ^ Габеллини Д., Д'Антона Дж., Моджио М. и др. (23 февраля 2006 г.). «Лице-лопаточно-плечевая мышечная дистрофия у мышей со сверхэкспрессией FRG1». Природа . 439 (7079): 973–977. Бибкод : 2006Natur.439..973G . дои : 10.1038/nature04422 . ПМИД 16341202 . S2CID 4427465 .
- ^ Клэпп Дж., Митчелл Л.М., Болланд Дж. и др. (август 2007 г.). «Эволюционная консервация кодирующей функции D4Z4, тандемного повтора ДНК, мутировавшего при лице-лопаточно-плечевой мышечной дистрофии» . Американский журнал генетики человека . 81 (2): 264–279. дои : 10.1086/519311 . ПМК 1950813 . ПМИД 17668377 .
- ^ Диксит М., Анссо Э., Тассин А. и др. (13 ноября 2007 г.). «DUX4, ген-кандидат лице-лопаточно-плечевой мышечной дистрофии, кодирует активатор транскрипции PITX1» . Труды Национальной академии наук США . 104 (46): 18157–18162. Бибкод : 2007PNAS..10418157D . дои : 10.1073/pnas.0708659104 . ПМК 2084313 . ПМИД 17984056 .
- ^ де Греф Дж.К., Леммерс Р.Дж., ван Энгелен Б.Г. и др. (октябрь 2009 г.). «Общие эпигенетические изменения D4Z4 при зависимом от сокращения и независимом от сокращения ЛЛПД». Человеческая мутация . 30 (10): 1449–1459. CiteSeerX 10.1.1.325.8388 . дои : 10.1002/humu.21091 . ПМИД 19728363 . S2CID 14517505 .
- ^ Снайдер Л., Асавачайчарн А., Тайлер А.Е. и др. (1 июля 2009 г.). «Транскрипты РНК, фрагменты размером с микроРНК и белки, полученные из единиц D4Z4: новые кандидаты в патофизиологию лице-лопаточно-плечевой дистрофии» . Молекулярная генетика человека . 18 (13): 2414–2430. дои : 10.1093/hmg/ddp180 . ПМК 2694690 . ПМИД 19359275 .
- ^ Перейти обратно: а б Колата Г (19 августа 2010 г.). «Обнаружено, что реанимированная «мусорная» ДНК вызывает болезни» . Нью-Йорк Таймс . Проверено 29 августа 2010 г.
- ^ Перейти обратно: а б «Вехи исследований, достигнутые исследованиями Общества ФСГ» . Общество ФШ . Архивировано из оригинала 23 августа 2010 г. Проверено 21 февраля 2024 г.
- ^ Сциони И., Греко Ф., Риччи Дж. и др. (6 апреля 2012 г.). «Крупномасштабный популяционный анализ бросает вызов современным критериям молекулярной диагностики фасцио-лопаточно-плечевой мышечной дистрофии» . Американский журнал генетики человека . 90 (4): 628–635. дои : 10.1016/j.ajhg.2012.02.019 . ПМЦ 3322229 . ПМИД 22482803 .
- ^ Гэн Л.Н., Яо З., Снайдер Л. и др. (17 января 2012 г.). «DUX4 активирует гены зародышевой линии, ретроэлементы и иммунные медиаторы: последствия лице-лопаточно-плечевой дистрофии» . Развивающая клетка . 22 (1): 38–51. дои : 10.1016/j.devcel.2011.11.013 . ПМК 3264808 . ПМИД 22209328 .
- ^ Джонс Т.И., Чен Дж.К., Рахимов Ф. и др. (15 октября 2012 г.). «Семейные исследования экспрессии DUX4 при лице-лопаточно-плечевой мышечной дистрофии: доказательства наличия модификаторов заболевания и количественная модель патогенеза» . Молекулярная генетика человека . 21 (20): 4419–4430. дои : 10.1093/hmg/dds284 . ПМЦ 3459465 . ПМИД 22798623 .
- ^ Кром Ю.Д., Тийссен П.Е., Янг Дж.М. и др. (апрель 2013 г.). «Внутренняя эпигенетическая регуляция макросателлитного повтора D4Z4 в модели трансгенных мышей при ЛЛПД» . ПЛОС Генетика . 9 (4): e1003415. дои : 10.1371/journal.pgen.1003415 . ПМК 3616921 . ПМИД 23593020 .
- ^ Ферребёф М., Мариот В., Бессьер Б. и др. (1 января 2014 г.). «Последующие гены-мишени DUX4 и DUX4 экспрессируются в мышцах FSHD плода» . Молекулярная генетика человека . 23 (1): 171–181. дои : 10.1093/hmg/ddt409 . ПМИД 23966205 .
- ^ Тавил Р., Макдермотт, член парламента, Пандия С., Кинг В., Киссел Дж., Менделл-младший, Григгс Р.К. (январь 1997 г.). «Пилотное исследование преднизолона при лице-лопаточно-плечевой мышечной дистрофии. Группа FSH-DY». Неврология . 48 (1): 46–9. дои : 10.1212/wnl.48.1.46 . ПМИД 9008492 . S2CID 729275 .
- ^ Киссел Дж.Т., Макдермотт, член парламента, Натараджан Р., Менделл Дж.Р., Пандия С., Кинг В.М., Григгс Р.К., Тавил Р. (май 1998 г.). «Пилотное исследование альбутерола при лице-лопаточно-плечевой мышечной дистрофии. Группа FSH-DY». Неврология . 50 (5): 1402–6. дои : 10.1212/wnl.50.5.1402 . ПМИД 9595995 . S2CID 24848310 .
- ^ Киссель Дж.Т., Макдермотт, член парламента, Менделл-младший, Кинг В.М., Пандия С., Григгс Р.К., Тавил Р., FSH-DY G (23 октября 2001 г.). «Рандомизированное двойное слепое плацебо-контролируемое исследование альбутерола при лице-лопаточно-плечевой дистрофии». Неврология . 57 (8): 1434–40. дои : 10.1212/wnl.57.8.1434 . ПМИД 11673585 . S2CID 28093111 .
- ^ ван дер Кои Э.Л., Фогельс О.Дж., ван Ассельдонк Р.Дж., Линдеман Э., Хендрикс Дж.К., Вольгемут М., ван дер Маарел С.М., Падберг Г.В. (24 августа 2004 г.). «Силовые тренировки и альбутерол при лице-лопаточно-плечевой мышечной дистрофии». Неврология . 63 (4): 702–8. дои : 10.1212/01.wnl.0000134660.30793.1f . ПМИД 15326246 . S2CID 22778327 .
- ^ Перейти обратно: а б Кэмпбелл А.Э., Олива Дж., Йейтс М.П., Чжун Дж.В., Шадл С.С., Снайдер Л., Сингх Н., Тай С., Хирамуки Ю., Тавил Р., ван дер Маарел С.М., Тапскотт С.Дж., Свердруп FM (4 сентября 2017 г.). «Ингибиторы бромодомена BET и агонисты бета-2-адренергических рецепторов, идентифицированные в ходе скрининга соединений, которые ингибируют экспрессию DUX4 в мышечных клетках ЛЛПД» . Скелетная мышца . 7 (1): 16. дои : 10.1186/s13395-017-0134-x . ПМЦ 5584331 . ПМИД 28870238 .
- ^ Эльшейх Б.Х., Боллман Э., Перуджа М., Кинг В., Галлоуэй Дж., Кисель Дж.Т. (24 апреля 2007 г.). «Пилотное исследование дилтиазема при лице-лопаточно-плечевой мышечной дистрофии». Неврология . 68 (17): 1428–9. дои : 10.1212/01.wnl.0000264017.08217.39 . ПМИД 17452589 . S2CID 361422 .
- ^ Wyeth начинает клинические испытания исследуемой терапии мышечной дистрофии MYO-029.
- ^ Малкольм Э. (18 июня 2019 г.). «АСЕ-083» . Новости мышечной дистрофии . Проверено 19 декабря 2019 г.
- ^ Вультаджио М (5 октября 2018 г.). «Почему Руфус Сьюэлл хотел сыграть злодея Джона Смита из «Человека в высоком замке»» . Newsweek . Архивировано из оригинала 6 ноября 2018 года . Проверено 12 апреля 2022 г.
- ^ Какутани М. (9 июня 2006 г.). « Стюарт: Жизнь наоборот», Александр Мастерс, Портрет бездомного» . Нью-Йорк Таймс . Архивировано из оригинала 16 января 2018 года.
- ^ Белый А (19 декабря 2023 г.). « Звезда «Рами» Стив Уэй участвует в инди-драме «Хорошие плохие дела» в качестве исполнительного продюсера (эксклюзивно)» . Голливудский репортер . Проверено 31 января 2024 г.
- ^ jkinoshita (25 августа 2022 г.). «В новом фильме будет показан мужчина с ЛЛПД» . Общество ФШД . Проверено 31 января 2024 г.
- ^ «Защитник инвалидов сохраняет свой голос с помощью iPhone» . Отдел новостей Apple . Проверено 21 февраля 2024 г.
- ^ Перейти обратно: а б Киносита Дж. (16 августа 2019 г.). «Что в имени? – Общество ФШД» . Общество ФШД . Архивировано из оригинала 12 апреля 2022 года . Проверено 18 августа 2019 г.
- ^ Перейти обратно: а б «Наша история» . Общество ФШД . Архивировано из оригинала 27 февраля 2022 года.
- ^ Перейти обратно: а б Бартлетт Дж. (19 августа 2014 г.). Бостонский деловой журнал https://web.archive.org/web/20220412220102/https://www.bizjournals.com/boston/blog/health-care/2014/08/dan-perez-living-with-and-fighting -против-a.html . Архивировано из оригинала 12 апреля 2022 года . Проверено 12 апреля 2022 г.
{{cite news}}: Отсутствует или пусто|title=( помощь ) - ^ «Общество ФШД» . НОРД (Национальная организация по редким заболеваниям) . Архивировано из оригинала 13 мая 2022 г. Проверено 12 апреля 2022 г.
- ^ «Общество FSHD получило аккредитацию от BBB Wise Giving Alliance» . Првеб . 26 апреля 2022 года. Архивировано из оригинала 29 апреля 2022 года . Проверено 29 апреля 2022 г.
- ^ «Это ФШД» . ХаффПост . 16 июля 2014 г. Архивировано из оригинала 2 августа 2019 г. . Проверено 12 апреля 2022 г.
- ^ Крамми С (30 марта 2017 г.). «Пара Киркланд собрала 3,2 миллиона долларов на исследование мышечной дистрофии ФСГ» . Киркланд Репортер . Архивировано из оригинала 16 июня 2017 года . Проверено 9 апреля 2022 г.
- ^ «О нас: Друзья исследований ФСГ» . Друзья исследования ФСГ . Архивировано из оригинала 17 февраля 2020 года . Проверено 9 апреля 2022 г.
- ^ AMRA Medical (13 октября 2021 г.). «Анализ МРТ всего тела компании AMRA Medical, использованный в исследовании сети клинических исследований ЛЛПД для разработки биомаркеров» . Cision PR Newswire . Архивировано из оригинала 1 ноября 2021 года . Проверено 9 апреля 2022 г.
- ^ Avidity Biosciences, Inc. (16 августа 2021 г.). «Avidity Biosciences вступает в сотрудничество с сетью клинических испытаний FSHD для поддержки разработки биомаркеров для будущего использования в клинических испытаниях» . Cision PR Newswire . Архивировано из оригинала 16 августа 2021 года.
- ^ Оверингтон С (24 сентября 2016 г.). «Он физически истощен, но его мозг острый. Бывший банкир Macquarie Билл Мосс снова в деле». Австралиец .
- ^ Перейти обратно: а б с Канцлер Дж. (7 сентября 2020 г.). «Покупатель нападает на кореллу Бретта Уайтли» . Австралиец . Проверено 9 апреля 2022 г.
- ^ Перейти обратно: а б Таскер SJ (26 мая 2018 г.). «Билл Мосс, целеустремленный биотехнолог и поиск лекарства» . Австралиец . Проверено 9 апреля 2022 г.
- ^ Перейти обратно: а б Фёрманс Н.К., Вриенс-Муньос Браво М., Падберг Г.В., Лафоре П., исследование семинара Европейской сети испытаний FSHD g (сентябрь 2021 г.). «Первый семинар Европейской сети судебных разбирательств FSHD: Работа над готовностью к судебным испытаниям по всей Европе» . Нервно-мышечные расстройства . 31 (9): 907–918. дои : 10.1016/j.nmd.2021.07.013 . hdl : 1887/3505452 . ПМИД 34404575 . S2CID 236217036 . Архивировано из оригинала 19 марта 2024 года.
- ^ Джадд А. (8 марта 2022 г.). «Основатель Lululemon Чип Уилсон жертвует 100 миллионов долларов на поиск лекарства от своей болезни спустя 30 лет после постановки диагноза» . Глобальные новости . Проверено 9 апреля 2022 г.
- ^ Изола Ф (23 апреля 2019 г.). « Ты не собираешься сдаваться»: шаг за шагом голос радио «Нетс» Крис Каррино продолжает идти вперед» . Атлетик . Архивировано из оригинала 14 февраля 2023 года . Проверено 9 апреля 2022 г.
- ^ «Актриса, преодолевающая барьеры, как первая главная актриса Бродвея в инвалидной коляске» . Новости Эн-Би-Си . 19 марта 2024 г. Проверено 21 февраля 2024 г.
- ^ «Профили сообщества: Актриса Мэдисон Феррис» . FSHD Откровенный разговор с Тимом Холленбеком . Аудиобум. 13 сентября 2022 г. Архивировано из оригинала 21 февраля 2024 г. Проверено 21 февраля 2024 г.
- ^ Шедлоски Д. (24 февраля 2020 г.). «Нет возможности остановить Моргана Хоффмана в его борьбе с мышечной дистрофией» . Гольф-дайджест . Архивировано из оригинала 5 марта 2022 года . Проверено 12 апреля 2022 г.
- ^ Сандомир Р. (3 февраля 2018 г.). «Доктор Арнольд Голд, 92 года, умер; сделал сострадательную заботу своим делом» . Нью-Йорк Таймс . ISSN 0362-4331 . Проверено 21 февраля 2024 г.
- ^ Рикард А., Петек Л., Миллер Д. (5 августа 2015 г.). «Эндогенная экспрессия DUX4 в мышечных трубках ЛЛПД достаточна, чтобы вызвать гибель клеток и нарушить пути сплайсинга РНК и миграции клеток» . Хм. Мол. Жене . 24 (20): 5901–14. дои : 10.1093/hmg/ddv315 . ПМЦ 4581613 . ПМИД 26246499 .
- ^ «Fulcrum Therapeutics приобретает глобальные права на Лосмапимод, потенциальную терапию, модифицирующую заболевание лице-лопаточно-плечевой мышечной дистрофии» . Биокосмос . Проверено 12 августа 2019 г.
- ^ «Результаты испытаний ReDUX4 превосходят ожидания» . Общество ФШД . Машина обратного пути. 24 июня 2021 г. Архивировано из оригинала 26 июня 2021 года . Проверено 26 июня 2021 г.
- ^ «Эффективность и безопасность лосмапимода у пациентов с лице-лопаточно-плечевой мышечной дистрофией (ЛЛПД)» . ClinicalTrials.gov . Национальная медицинская библиотека США . Проверено 12 августа 2019 г.
- ^ «Фасио представит на Конгрессе Всемирного общества мышц» . Терапия для лица . 30 сентября 2019 года . Проверено 9 ноября 2019 г.
- ^ «Фасио раскрывает новый механизм, направленный на причину ЛЛПД» . Терапия для лица . 24 июня 2019 г. Проверено 9 ноября 2019 г.
- ^ «Arrowhead Pharmaceuticals объявляет кандидата на лекарство от ЛЛПД» . Общество ФШД . 15 апреля 2021 г. Проверено 15 апреля 2021 г.
- ^ «Arrowhead объявляет ARO-DUX4 как первый кандидат на РНКи, нацеленный на мышцы, использующий платформу TRiMTM» . Бизнесвайр . 15 апреля 2021 г. Проверено 15 апреля 2021 г.
- ^ Саад Нью-Йорк, Аль-Харсан М., Гарвик-Коппенс С.Э., Чермахини Г.А., Харпер М.А., Пало А., Будро Р.Л., Харпер С.К. (декабрь 2021 г.). «Человеческая микроРНК-миР-675 ингибирует экспрессию DUX4 и может быть использована в качестве потенциального средства лечения лице-лопаточно-плечевой мышечной дистрофии» . Природные коммуникации . 12 (1): 7128. Бибкод : 2021NatCo..12.7128S . дои : 10.1038/ s41467-021-27430-1 ПМЦ 8654987 . ПМИД 34880230 .
- ^ Жубер Р., Марио В., Шарпантье М., Конкорде Ж.П., Дюмонсо Ж. (23 декабря 2020 г.). «Редактирование генов, направленное на сигнал полиаденилирования DUX4: терапия ЛЛПД?» . Журнал персонализированной медицины . 11 (1): 7. дои : 10.3390/jpm11010007 . ПМЦ 7822190 . ПМИД 33374516 .
- ^ Дезимоун А.М., Лешик Дж., Вагнер К., Эмерсон К.П. (11 декабря 2019 г.). «Идентификация пути гиалуроновой кислоты как терапевтической мишени при лице-лопаточно-плечевой мышечной дистрофии» . Достижения науки . 5 (12): eaaw7099. Бибкод : 2019SciA....5.7099D . дои : 10.1126/sciadv.aaw7099 . ПМК 6905861 . ПМИД 31844661 .
- ^ Боснаковски Д., да Силва М.Т., Санни С.Т., Энер Э.Т., Тосо Е.А., Юань С., Цуй З., Уолтерс М.А., Джадхав А., Киба М. (сентябрь 2019 г.). «Новый ингибитор P300 обращает вспять опосредованное DUX4 глобальное гиперацетилирование гистона H3, экспрессию целевого гена и гибель клеток» . Достижения науки . 5 (9): eaaw7 Бибкод : 2019SciA.... 5.7781B дои : 10.1126/sciadv.aaw7781 . ПМК 6739093 . ПМИД 31535023 .
- ^ Пассерье Э., Айо М., Жоссент А., Карнак Дж., Гузи Ф., Пиллард Ф., Пико MC, Бёккер К., Хьюгон Дж., Пинсмаил Дж., Дефрен Ж.О., Веррипс Т., Мерсье Дж., Лаудж-Шенивесс Д. (апрель 2015 г.). «Влияние витамина С, витамина Е, глюконата цинка и добавок селенометионина на мышечную функцию и биомаркеры окислительного стресса у пациентов с лице-лопаточно-плечевой дистрофией: двойное слепое рандомизированное контролируемое клиническое исследование» (PDF) . Свободно-радикальная биология и медицина . 81 : 158–69. doi : 10.1016/j.freeradbiomed.2014.09.014 . ПМИД 25246239 . S2CID 10971952 .
- ^ Хан Дж.Дж., Курилло Г., Абреш Р.Т., де Бие Э., Никоричи А., Байчи Р. (февраль 2015 г.). «Доступное рабочее пространство при лице-лопаточно-плечевой мышечной дистрофии (ЛЛПД) от Kinect» . Мышцы и нервы . 51 (2): 168–175. дои : 10.1002/mus.24287 . ISSN 0148-639X . ПМК 4233016 . ПМИД 24828906 .
- ^ Дэни А, Барбе С, Рапен А, Ревейер С, Ардуэн ЖБ, Моррон И, Волак-Тьерри А, Драме М, Калмус А, Саккони С, Бассес Г, Тиффро В, Ришар И, Галле Б, Приджент Х, Тайар Р, Джолли Д., Новелла Дж.Л., Бойер ФК (2015). «Построение опросника качества жизни при медленно прогрессирующих нервно-мышечных заболеваниях». Исследование качества жизни . 24 (11): 2615–2623. дои : 10.1007/s11136-015-1013-8 . ISSN 0962-9343 . ПМИД 26141500 . S2CID 25834947 .
- ^ Мул К., Хамаде Т., Хорлингс К., Тавил Р., Стэтленд Дж.М., Саккони С., Корбетт А.Дж., Воерманс Н.К., Фабер К.Г., ван Энгелен Б., Меркис I (10 апреля 2021 г.). «Шкала общей инвалидности FacioScapuloHumeral при мышечной дистрофии, построенная Рашем (FSHD-RODS)» . Европейский журнал неврологии . 28 (7): 2339–2348. дои : 10.1111/ene.14863 . ПМЦ 8251612 . ПМИД 33838063 .
- ^ Эйхингер К., Хитволе С., Иядурай С., Кинг В., Бейкер Л., Хайнингер С., Бартлетт А., Дилек Н., Мартенс В.Б., Макдермотт М., Киссель Дж.Т., Тавил Р., Стэтленд Дж.М. (30 января 2018 г.). «Комплексный функциональный показатель исхода лице-лопаточно-плечевой мышечной дистрофии» . Мышцы и нервы . 58 : 72–78. дои : 10.1002/mus.26088 . ПМК 6066464 . ПМИД 29381807 .
Внешние ссылки
[ редактировать ]- Всемирный альянс ЛЛПД: организации по защите прав пациентов с ЛЛПД по всему миру.
- список клинических исследований ЛЛПД , Clinicaltrials.gov .
- ФШ в NIH / UW GeneTests