Нейродегенеративное заболевание
| Нейродегенеративное заболевание | |
|---|---|
 | |
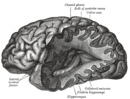
| Нормальный мозг слева контрастирует со структурными изменениями, показанными в мозге справа человека с болезнью Альцгеймера , наиболее распространенным нейродегенеративным заболеванием. [1] | |
| Специальность | Неврология , Психиатрия |
Нейродегенеративное заболевание вызвано прогрессирующей гибелью нейронов в процессе, известном как нейродегенерация . [2] [3] Повреждение нейронов также может в конечном итоге привести к их гибели . Нейродегенеративные заболевания включают боковой амиотрофический склероз , рассеянный склероз , болезнь Паркинсона , болезнь Альцгеймера , болезнь Хантингтона , множественную системную атрофию , тауопатии и прионные заболевания . Нейродегенерацию можно обнаружить в мозге на многих различных уровнях нейронных сетей, от молекулярного до системного. Поскольку не существует известного способа обратить вспять прогрессирующую дегенерацию нейронов, эти заболевания считаются неизлечимыми; однако исследования показали, что двумя основными факторами, способствующими нейродегенерации, являются окислительный стресс и воспаление. [4] [5] [6] [7] Биомедицинские исследования выявили множество сходств между этими заболеваниями на субклеточном уровне, включая атипичные белковые сборки (например, протеинопатию ) и индуцированную гибель клеток. [8] [9] Эти сходства позволяют предположить, что терапевтические достижения в борьбе с одним нейродегенеративным заболеванием могут улучшить и другие заболевания.
По оценкам, что касается нейродегенеративных заболеваний, в 2019 году во всем мире деменцией страдали 55 миллионов человек , а к 2050 году эта цифра увеличится до 139 миллионов человек. [10]
Специфические расстройства
[ редактировать ]Последствия нейродегенерации могут широко варьироваться в зависимости от конкретной пораженной области: от проблем, связанных с движением, до развития деменции. [11] [12]
болезнь Альцгеймера
[ редактировать ]
Болезнь Альцгеймера (БА) — хроническое нейродегенеративное заболевание, приводящее к потере нейронов и синапсов в коре головного мозга и некоторых подкорковых структурах, что приводит к грубой атрофии височной доли , теменной доли , а также частей лобной коры и поясной извилины . [13] Это самое распространенное нейродегенеративное заболевание. [1] Даже несмотря на то, что на поиски лечения болезни Альцгеймера были потрачены миллиарды долларов, эффективных методов лечения найдено не было. [14] В клинических исследованиях стабильные и эффективные стратегии лечения БА имеют частоту неудач в 99,5%. [15] Причины такого уровня неудач включают неподходящие дозы лекарств, неправильный выбор целей и участников, а также недостаточное знание патофизиологии БА. В настоящее время диагностика болезни Альцгеймера находится на низком уровне, и необходимо использовать более совершенные методы для различных аспектов клинической диагностики. [16] Болезнь Альцгеймера имеет 20% ошибочный диагноз. [16]
Патология БА прежде всего характеризуется наличием амилоидных бляшек и нейрофибриллярных клубков . Бляшки состоят из небольших пептидов , обычно длиной 39–43 аминокислоты, называемых бета-амилоидом (также обозначаемым как A-бета или Aβ). Бета-амилоид представляет собой фрагмент более крупного белка, называемого белком-предшественником амилоида (APP), трансмембранного белка , который проникает через мембрану нейрона. АРР, по-видимому, играет роль в нормальном росте нейронов, выживании и восстановлении после травмы. [17] [18] АРР расщепляется на более мелкие фрагменты такими ферментами , как гамма-секретаза и бета-секретаза . [19] Один из этих фрагментов дает начало фибриллам бета-амилоида, которые могут самоорганизовываться в плотные внеклеточные амилоидные бляшки. [20] [21]
болезнь Паркинсона
[ редактировать ]Болезнь Паркинсона (БП) является вторым по распространенности нейродегенеративным заболеванием. [22] Обычно это проявляется брадикинезией , ригидностью, тремором покоя и нестабильностью позы. Сообщается, что общий уровень распространенности БП колеблется от 15 на 100 000 до 12 500 на 100 000, а заболеваемость БП - от 15 на 100 000 до 328 на 100 000, при этом заболевание менее распространено в азиатских странах.
БП прежде всего характеризуется гибелью дофаминергических нейронов в черной субстанции , области среднего мозга . Причина этой избирательной гибели клеток неизвестна. Примечательно, что комплексы и агрегаты альфа-синуклеин - убиквитин накапливаются в тельцах Леви внутри пораженных нейронов. Считается, что дефекты механизмов и регуляции транспорта белков, таких как RAB1 , могут играть роль в механизме этого заболевания. [23] Нарушение аксонального транспорта альфа-синуклеина также может приводить к его накоплению в тельцах Леви. Эксперименты выявили снижение скорости транспорта как дикого типа, так и двух мутантных альфа-синуклеинов, связанных с семейной болезнью Паркинсона, через аксоны культивируемых нейронов. [24] Повреждение мембран альфа-синуклеином может быть еще одним механизмом болезни Паркинсона. [25]
Основным известным фактором риска является возраст. Мутации в таких генах, как α-синуклеин (SNCA), киназа 2 с богатыми лейцином повторами (LRRK2), глюкоцереброзидаза (GBA) и тау-белок (MAPT), также могут вызывать наследственную БП или повышать риск БП. [26] Хотя БП является вторым по распространенности нейродегенеративным заболеванием, проблемы с диагностикой все еще сохраняются. [27] Проблемы с обонянием — широко распространенный симптом болезни Паркинсона (БП), однако некоторые неврологи ставят под сомнение его эффективность. [27] Этот метод оценки является источником разногласий среди медицинских работников. [27] Микробиом кишечника может играть роль в диагностике БП, и исследования предлагают различные способы, которые могут революционизировать будущее лечения БП. [28]
болезнь Хантингтона
[ редактировать ]Болезнь Хантингтона (БГ) — редкое аутосомно-доминантное нейродегенеративное заболевание, обусловленное мутациями гена хантингтина (HTT) . HD характеризуется потерей средних шипистых нейронов и астроглиозом . [29] [30] [31] Первой областью мозга, которая подвергается существенному поражению, является полосатое тело , за которым следует дегенерация лобной и височной коры. [32] полосатого тела Субталамические ядра посылают управляющие сигналы бледному шару , который инициирует и модулирует движение. Таким образом, более слабые сигналы от субталамических ядер вызывают снижение инициации и модуляции движений, что приводит к характерным для расстройства движениям, особенно к хорее . [33] Болезнь Хантингтона проявляется в более позднем возрасте, хотя белки, вызывающие заболевание, способствуют проявлению заболевания уже на ранних стадиях у людей, пораженных этими белками. [34] Помимо того, что HD является нейродегенеративным расстройством, он связан с проблемами нервного развития. [34]
HD вызывается расширением полиглутаминового тракта в гене хантингтина, что приводит к образованию мутантного хантингтина. Агрегаты мутантного хантингтина образуются в нейронах в виде телец включения и могут быть непосредственно токсичными. Кроме того, они могут повреждать молекулярные моторы и микротрубочки, нарушая нормальный аксональный транспорт , что приводит к нарушению транспорта важных грузов, таких как BDNF . [24] Болезнь Хантингтона в настоящее время не имеет эффективных методов лечения, которые могли бы изменить течение заболевания. [35]
Рассеянный склероз
[ редактировать ]Рассеянный склероз (РС) — хроническое изнурительное демиелинизирующее заболевание центральной нервной системы , вызванное аутоиммунной атакой, приводящей к прогрессирующей потере миелиновой оболочки аксонов нейронов. [36] Результирующее снижение скорости передачи сигнала приводит к потере функциональности, которая включает как когнитивные, так и двигательные нарушения в зависимости от локализации поражения. [36] Прогрессирование рассеянного склероза происходит из-за эпизодов усиления воспаления, которое, как предполагается, происходит из-за высвобождения антигенов, таких как гликопротеин миелин-олигодендроцитов , основной белок миелина и протеолипидный белок , вызывающих аутоиммунный ответ. [37] Это запускает каскад сигнальных молекул, которые заставляют Т-клетки, В-клетки и макрофаги преодолевать гематоэнцефалический барьер и атаковать миелин на аксонах нейронов, что приводит к воспалению. [38] Дальнейшее высвобождение антигенов приводит к последующей дегенерации, вызывающей усиление воспаления. [39] Рассеянный склероз представляет собой спектр, основанный на степени воспаления: у большинства пациентов наблюдаются ранние рецидивирующие и ремиттирующие эпизоды разрушения нейронов после периода выздоровления. У некоторых из этих людей может произойти переход к более линейному прогрессированию заболевания, в то время как около 15% других начинаются с прогрессирующего течения в начале рассеянного склероза. Воспалительная реакция способствует потере серого вещества, и в результате современная литература посвящена борьбе с аутовоспалительным аспектом заболевания. [38] Хотя существует несколько предполагаемых причинно-следственных связей между EBV и аллелем HLA-DRB1*15:01 с возникновением рассеянного склероза (они могут способствовать степени аутоиммунной атаки и возникающему в результате воспаления), они не определяют начало рассеянного склероза. [38]
Боковой амиотрофический склероз
[ редактировать ]Боковой амиотрофический склероз (БАС), обычно называемый болезнью Лу Герига , представляет собой редкое нейродегенеративное заболевание, характеризующееся постепенной потерей как верхних мотонейронов (ВМН), так и нижних мотонейронов (ЛМН). [40] Хотя первоначальные симптомы могут различаться, у большинства пациентов развивается слабость скелетных мышц, которая прогрессирует и охватывает все тело. [40] Точная этиология БАС остается неизвестной. В 1993 году миссенс-мутации в гене, кодирующем антиоксидантный фермент супероксиддисмутазу 1 (СОД1), были обнаружены у пациентов с семейным БАС. Совсем недавно ДНК-связывающий белок TAR 43 (TDP-43) и белковые агрегаты, слитые в саркоме (FUS) в некоторых случаях заболевания были вовлечены , а мутация в хромосоме 9 ( C9orf72 ) считается наиболее распространенной из известных. причина спорадического БАС. Ранняя диагностика БАС сложнее, чем других нейродегенеративных заболеваний, поскольку не существует высокоэффективных способов определения его раннего начала. [40] В настоящее время проводятся исследования по диагностике БАС с помощью тестов верхних мотонейронов. [41] Шкала верхних двигательных нейронов Пенна (PUMNS) состоит из 28 критериев с диапазоном оценок от 0 до 32. [41] Более высокий балл указывает на более высокий уровень нагрузки на верхние мотонейроны. [41] PUMNS оказался весьма эффективным при определении нагрузки на верхние мотонейроны у больных пациентов. [41]
Независимые исследования in vitro предоставили доказательства того, что основные клеточные участки, где действуют мутации SOD1, расположены на астроцитах . [42] [43] Астроциты затем оказывают токсическое воздействие на мотонейроны . Конкретный механизм токсичности еще предстоит изучить, но полученные результаты важны, поскольку они указывают на то, что в нейродегенерации участвуют не только нейронные клетки, но и другие клетки. [44]
Болезнь Баттена
[ редактировать ]Болезнь Баттена — редкое и смертельное рецессивное нейродегенеративное заболевание, которое начинается в детстве. [45] Болезнь Баттена – это общее название группы лизосомальных нарушений накопления, известных как нейрональные цероидные липофусцинозы (НКЛ), каждое из которых вызвано конкретной мутацией гена. [45] из них тринадцать. [46] Поскольку болезнь Баттена встречается довольно редко, ее распространенность во всем мире составляет примерно 1 случай на 100 000 живорождений. [46] В Северной Америке болезнь NCL3 (ювенильный NCL) обычно проявляется в возрасте от 4 до 7 лет. [47] Болезнь Баттена характеризуется двигательными нарушениями, эпилепсией , деменцией , потерей зрения и сокращением продолжительности жизни. [48] Потеря зрения является частым первым признаком болезни Баттена. [47] Потере зрения обычно предшествуют когнитивные и поведенческие изменения, судороги и потеря способности ходить. [47] По мере прогрессирования заболевания у людей часто возникают сердечные аритмии и трудности с приемом пищи. [47] Диагностика болезни Баттена зависит от совокупности многих критериев: клинических признаков и симптомов, оценки состояния глаз, результатов электроэнцефалограммы (ЭЭГ) и магнитно-резонансной томографии головного мозга (МРТ). [46] Диагноз, поставленный на основании этих результатов, подтверждается генетическими и биохимическими исследованиями. [46] До последних нескольких лет не существовало эффективных методов лечения, которые могли бы предотвратить широкое распространение заболевания. [46] В последние годы было создано больше моделей для ускорения процесса исследования методов лечения болезни Баттена. [46]
Болезнь Крейтцфельдта-Якоба
[ редактировать ]Болезнь Крейтцфельдта-Якоба (БКЯ) — прионное заболевание, характеризующееся быстропрогрессирующей деменцией. [49] Неправильно свернутые белки, называемые прионами, агрегируются в тканях мозга, что приводит к гибели нервных клеток. [50] Вариант болезни Крейтцфельдта-Якоба (вБКЯ) — это инфекционная форма, которая возникает из мяса коровы, инфицированной губчатой энцефалопатией крупного рогатого скота , также называемой коровьим бешенством. [51]
Факторы риска
[ редактировать ]Старение
[ редактировать ]Самым большим фактором риска нейродегенеративных заболеваний является старение . Мутации митохондриальной ДНК , а также окислительный стресс способствуют старению. [52] Многие из этих заболеваний возникают поздно, то есть существует некоторый фактор, который меняется с возрастом человека для каждого заболевания. [8] Одним из постоянных факторов является то, что при каждом заболевании нейроны постепенно теряют функцию по мере прогрессирования заболевания с возрастом. Было высказано предположение, что накопление повреждений ДНК обеспечивает основную причинную связь между старением и нейродегенеративными заболеваниями. [53] [54] Около 20–40% здоровых людей в возрасте от 60 до 78 лет испытывают заметное снижение когнитивных функций в нескольких областях, включая рабочую, пространственную и эпизодическую память, а также скорость обработки информации. [55]
Инфекции
[ редактировать ]
Исследование с использованием электронных медицинских записей показывает, что 45 (22 из них воспроизведены в Биобанке Великобритании ) воздействия вируса могут значительно повысить риск нейродегенеративных заболеваний, в том числе в течение 15 лет после заражения. [56] [57]
Механизмы
[ редактировать ]Генетика
[ редактировать ]Многие нейродегенеративные заболевания вызваны генетическими мутациями , большинство из которых расположены в совершенно неродственных генах. При многих различных заболеваниях мутировавший ген имеет общую особенность: повтор триплета нуклеотидов CAG. CAG кодирует аминокислоту глутамин . Повторение CAG приводит к образованию полиглутаминового (polyQ) тракта . Заболевания, связанные с такими мутациями, известны как нарушения тринуклеотидных повторов . [58] [59]
Полиглутаминовые повторы обычно вызывают доминантный патогенез. Дополнительные остатки глутамина могут приобретать токсичные свойства различными способами, включая нерегулярные пути сворачивания и деградации белков, изменение субклеточной локализации и аномальные взаимодействия с другими клеточными белками. [58] В исследованиях PolyQ часто используются различные модели на животных, поскольку существует четко определенный триггер – расширение повторов. Обширные исследования были проведены с использованием моделей нематод ) ( C. elegans ) и плодовой мухи ( дрозофилы , мышей и приматов, не являющихся человеком. [59] [60]
Девять наследственных нейродегенеративных заболеваний вызваны экспансией тринуклеотида CAG и полиQ-тракта, включая болезнь Хантингтона и спиноцеребеллярную атаксию . [61]
Эпигенетика
[ редактировать ]При этом типе патологии показано наличие эпигенетических модификаций некоторых генов. Примером может служить ген FKBP5 , экспрессия которого постепенно увеличивается с возрастом и связана со стадией по Брааку и увеличением тау-патологии как in vitro, так и на мышиных моделях AD. [62]
Неправильное сворачивание белка
[ редактировать ]Некоторые нейродегенеративные заболевания классифицируются как протеопатии , поскольку они связаны с агрегацией неправильно свернутых белков . Белковая токсичность является одним из ключевых механизмов многих нейродегенеративных заболеваний. [63]
- альфа-синуклеин : может агрегироваться с образованием нерастворимых фибрилл при патологических состояниях, характеризующихся тельцами Леви , таких как болезнь Паркинсона, деменция с тельцами Леви и множественная системная атрофия . Альфа-синуклеин является основным структурным компонентом фибрилл телец Леви. Кроме того, фрагмент альфа-синуклеина, известный как неабета-компонент (NAC), обнаруживается в амилоидных бляшках при болезни Альцгеймера .
- тау : гиперфосфорилированный тау-белок является основным компонентом нейрофибриллярных клубков при болезни Альцгеймера; Тау-фибриллы являются основным компонентом телец Пика, обнаруженных при поведенческом варианте лобно-височной деменции .
- бета-амилоид : основной компонент амилоидных бляшек при болезни Альцгеймера.
- Прион : основной компонент прионных заболеваний и трансмиссивной губчатой энцефалопатии .
Внутриклеточные механизмы
[ редактировать ]Пути деградации белка
[ редактировать ]Болезнь Паркинсона и болезнь Хантингтона возникают с поздним началом и связаны с накоплением внутриклеточных токсичных белков. Заболевания, вызванные агрегацией белков, известны как протеопатии , и они в первую очередь вызываются агрегатами в следующих структурах: [8]
- цитозоль, например болезнь Паркинсона и Хантингтона
- ядро, например, спиноцеребеллярная атаксия типа 1 (SCA1)
- эндоплазматический ретикулум (ЭР) (как видно из мутаций нейросерпина, которые вызывают семейную энцефалопатию с тельцами включения нейросерпина)
- внеклеточно экскретируемые белки, бета-амилоид при болезни Альцгеймера
Эукариотические клетки используют два основных способа удаления проблемных белков или органелл:
- убиквитин-протеасома: белок убиквитин наряду с ферментами играет ключевую роль в деградации многих белков, вызывающих протеопатии, включая экспансию полиQ и альфа-синуклеины. Исследования показывают, что протеасомные ферменты, возможно, не смогут правильно расщеплять эти нерегулярные белки, что может привести к появлению более токсичных видов. Это основной путь, по которому клетки расщепляют белки. [8]
- Снижение активности протеасом согласуется с моделями формирования внутриклеточных белковых агрегатов. До сих пор неизвестно, являются ли эти агрегаты причиной или результатом нейродегенерации. [8]
- Путь аутофагия-лизосома: форма запрограммированной гибели клеток (PCD). Этот путь становится благоприятным, когда белок склонен к агрегации, что означает, что он является плохим субстратом протеасом. Это можно разделить на две формы аутофагии: макроаутофагию и шаперон-опосредованную аутофагию (CMA) . [8]
- макроаутофагия участвует в рециркуляции питательных веществ макромолекул в условиях голодания, определенных путях апоптоза, а при ее отсутствии приводит к образованию убиквинированных включений. В экспериментах на мышах с нокаутом генов макроаутофагии, ограниченных нейронами, развиваются внутринейрональные агрегаты, приводящие к нейродегенерации. [8]
- Дефекты аутофагии, опосредованные шаперонами, также могут приводить к нейродегенерации. Исследования показали, что мутантные белки связываются с рецепторами пути CMA на лизосомальной мембране и тем самым блокируют свою собственную деградацию, а также деградацию других субстратов. [8]
Повреждение мембраны
[ редактировать ]Повреждение мембран органелл мономерными или олигомерными белками также может способствовать этим заболеваниям. Альфа-синуклеин может повреждать мембраны, вызывая их искривление. [25] и вызывают обширную тубуляцию и везикуляцию при инкубации с искусственными фосфолипидными везикулами. [25] Трубочки, образующиеся из этих липидных везикул, состоят как из мицеллярных, так и из двухслойных трубок. Обширная индукция искривления мембраны вредна для клетки и в конечном итоге приводит к ее гибели. Помимо трубчатых структур, альфа-синуклеин также может образовывать липопротеиновые наночастицы, подобные аполипопротеинам.
Митохондриальная дисфункция
[ редактировать ]Наиболее распространенной формой гибели клеток при нейродегенерации является внутренний путь апоптоза митохондрий. Этот путь контролирует активацию каспазы-9, регулируя высвобождение цитохрома с из митохондриального межмембранного пространства . Активные формы кислорода (АФК) являются нормальными побочными продуктами активности дыхательной цепи митохондрий. Концентрация АФК опосредована митохондриальными антиоксидантами, такими как супероксиддисмутаза марганца (СОД2) и глутатионпероксидаза . Чрезмерное производство АФК ( окислительный стресс ) является центральным признаком всех нейродегенеративных заболеваний. Помимо генерации АФК, митохондрии также участвуют в функциях жизнеобеспечения, включая гомеостаз кальция, PCD, деление и слияние митохондрий , концентрацию липидов в митохондриальных мембранах и переход проницаемости митохондрий. Митохондриальные заболевания , приводящие к нейродегенерации, вероятно, по крайней мере на каком-то уровне, затрагивают все эти функции. [64]
Имеются убедительные доказательства того, что митохондриальная дисфункция и окислительный стресс играют причинную роль в патогенезе нейродегенеративных заболеваний, в том числе в четырех наиболее известных заболеваниях: болезни Альцгеймера , Паркинсона , Хантингтона и боковом амиотрофическом склерозе . [52]
Нейроны особенно уязвимы к окислительному повреждению из-за их сильной метаболической активности, связанной с высоким уровнем транскрипции , высоким потреблением кислорода и слабой антиоксидантной защитой. [65] [66]
повреждение ДНК
[ редактировать ]Мозг метаболизирует пятую часть потребляемого кислорода, а активные формы кислорода, образующиеся в результате окислительного метаболизма, являются основным источником повреждения ДНК в мозге . клетки Повреждение ДНК особенно вредно, поскольку ДНК является основой для производства белка и, в отличие от других молекул, ее нельзя просто заменить путем повторного синтеза. Уязвимость постмитотических нейронов к повреждениям ДНК (таким как окислительные повреждения или определенные типы разрывов цепей ДНК) в сочетании с постепенным снижением активности механизмов репарации может привести к накоплению повреждений ДНК с возрастом и способствовать старению мозга. и нейродегенерация. [67] Одноцепочечные разрывы ДНК встречаются часто и связаны с нейродегенеративным заболеванием атаксия- глазодвигательная апраксия . [68] [66] Повышенное окислительное повреждение ДНК в головном мозге связано с болезнью Альцгеймера и болезнью Паркинсона . [68] Дефектная репарация ДНК связана с нейродегенеративными заболеваниями, такими как болезнь Альцгеймера, боковой амиотрофический склероз , атаксия-телеангиэктазия , синдром Кокейна , болезнь Паркинсона и пигментная ксеродермия . [68] [67]
Аксональный транспорт
[ редактировать ]Аксональный отек и аксональные сфероиды наблюдаются при многих различных нейродегенеративных заболеваниях. Это говорит о том, что дефектные аксоны не только присутствуют в больных нейронах, но также могут вызывать определенные патологические повреждения из-за накопления органелл. Аксональный транспорт может быть нарушен различными механизмами, включая повреждение кинезина и цитоплазматического динеина , микротрубочек , грузов и митохондрий . [24] Когда аксональный транспорт серьезно нарушается, дегенеративный путь, известный как валлериоподобная дегенерация . часто запускается [69]
Запрограммированная гибель клеток
[ редактировать ]Программируемая клеточная смерть (ПКС) — это смерть клетки в любой форме, опосредованная внутриклеточной программой. [70] Этот процесс может активироваться при нейродегенеративных заболеваниях, включая болезнь Паркинсона, боковой амитрофический склероз, болезнь Альцгеймера и болезнь Хантингтона. [71] ПКС, наблюдаемая при нейродегенеративных заболеваниях, может быть непосредственно патогенной; альтернативно, PCD может возникнуть в ответ на другие травмы или болезненные процессы. [9]
Апоптоз (тип I)
[ редактировать ]Апоптоз — это форма запрограммированной гибели клеток в многоклеточных организмах. Это один из основных типов запрограммированной гибели клеток (ПКС), который включает в себя серию биохимических событий, приводящих к характерной морфологии клеток и их гибели.
- Внешние пути апоптоза: возникают, когда факторы вне клетки активируют рецепторы гибели клеточной поверхности (например, Fas), что приводит к активации каспаз -8 или -10. [9]
- Внутренние пути апоптоза: возникают в результате высвобождения митохондриями цитохрома С или нарушений эндоплазматического ретикулума, каждый из которых приводит к активации каспазы-9. Ядро — это другие и аппарат Гольджи органеллы, имеющие датчики повреждения, которые могут вести клетки по апоптотическим путям. [9] [72]
Каспазы (протеазы цистеин-аспарагиновой кислоты) расщепляют очень специфические аминокислотные остатки. Различают два типа каспаз: инициаторы и эффекторы . Инициаторные каспазы расщепляют неактивные формы эффекторных каспаз. Это активирует эффекторы, которые, в свою очередь, расщепляют другие белки, что приводит к инициации апоптоза. [9]
Аутофагия (тип II)
[ редактировать ]Аутофагия — это форма внутриклеточного фагоцитоза , при которой клетка активно потребляет поврежденные органеллы или неправильно свернутые белки, инкапсулируя их в аутофагосому , которая сливается с лизосомой, разрушая содержимое аутофагосомы. Поскольку при многих нейродегенеративных заболеваниях наблюдаются необычные белковые агрегаты, предполагается, что дефекты аутофагии могут быть распространенным механизмом нейродегенерации. [9]
Цитоплазматический (тип III)
[ редактировать ]PCD также может возникать в результате неапоптотических процессов, также известных как тип III или гибель цитоплазматических клеток. Например, ПКС III типа может быть вызвана трофотоксичностью или гиперактивацией рецепторов трофических факторов. Цитотоксины, индуцирующие ПКС, могут вызывать некроз при низких концентрациях или апонекроз (сочетание апоптоза и некроза) при более высоких концентрациях. До сих пор неясно, какая именно комбинация апоптоза, неапоптоза и некроза вызывает различные виды апонекроза. [9]
Трансглутаминаза
[ редактировать ]Трансглутаминазы — это человеческие ферменты , повсеместно присутствующие в организме человека и, в частности, в мозге. [73]
Основная функция трансглутаминаз — связывать белки и пептиды внутри- и межмолекулярно с помощью ковалентных связей, называемых изопептидными связями , в реакции, называемой трансамидированием или сшивкой . [73]
этих белков и пептидов трансглутаминазой Связывание заставляет их слипаться. Полученные структуры становятся чрезвычайно устойчивыми к химическому и механическому разрушению. [73]
Большинство важных нейродегенеративных заболеваний человека имеют свойство иметь аномальные структуры, состоящие из белков и пептидов . [73]
Каждое из этих нейродегенеративных заболеваний имеет один (или несколько) специфический основной белок или пептид. При болезни Альцгеймера это бета-амилоид и тау . При болезни Паркинсона это альфа-синуклеин . При болезни Гентингтона это хантингтин . [73]
трансглутаминазы Субстраты : бета-амилоид , тау , альфа-синуклеин и хантингтин, было доказано, являются субстратами трансглутаминаз как in vitro или in vivo, то есть они могут связываться трасглутаминазами ковалентными связями друг с другом и потенциально с любым другим субстратом трансглутаминазы. в мозгу. [73]
Расширенная экспрессия трансглутаминазы : Доказано, что при этих нейродегенеративных заболеваниях (болезнях Альцгеймера, Паркинсона и Хантингтона) фермента трансглутаминазы . повышается экспрессия [73]
Наличие изопептидных связей в этих структурах: Наличие изопептидных связей (результат трансглутаминазной реакции) обнаружено в аномальных структурах, характерных для этих нейродегенеративных заболеваний . [73]
Совместная локализация: Совместная локализация изопептидных связей, опосредованных трансглутаминазой , с этими аномальными структурами была обнаружена при аутопсии головного мозга пациентов с этими заболеваниями. [73]
Управление
[ редактировать ]Процесс нейродегенерации недостаточно изучен, поэтому болезни, вызванные им, пока неизлечимы.
Модели животных в исследованиях
[ редактировать ]В поисках эффективных методов лечения (в отличие от паллиативной помощи ) исследователи используют модели болезней на животных для тестирования потенциальных терапевтических средств. Модельные организмы предоставляют недорогие и относительно быстрые средства для выполнения двух основных функций: идентификации цели и проверки цели. В совокупности они помогают показать ценность любых конкретных терапевтических стратегий и лекарств при попытке облегчить тяжесть заболевания. Примером может служить препарат Димебон от компании Medivation, Inc. В 2009 году этот препарат проходил III фазу клинических испытаний по применению при болезни Альцгеймера, а также II фазу клинических испытаний по применению при болезни Хантингтона. [59] В марте 2010 года были опубликованы результаты III фазы клинических испытаний; Исследуемый препарат для лечения болезни Альцгеймера «Димебон» потерпел неудачу в ключевом исследовании CONNECTION у пациентов с легкой и умеренной формой заболевания. [74] Благодаря CONCERT оставшееся исследование фазы III Pfizer и Medivation по димебону (латрепирдину) при болезни Альцгеймера в 2012 году потерпело неудачу, что фактически положило конец развитию этого показания. [75]
В другом эксперименте с использованием крысиной модели болезни Альцгеймера было продемонстрировано, что системное введение гипоталамического богатого пролином пептида (PRP)-1 оказывает нейропротекторное действие и может предотвратить нейродегенерацию бета-амилоида 25–35 гиппокампа. Это говорит о том, что PRP-1 может иметь терапевтическую ценность. [76]
Другие направления расследования
[ редактировать ]Деградация белков предлагает терапевтические возможности как для предотвращения синтеза, так и для деградации нерегулярных белков. Также существует интерес к усилению аутофагии, чтобы помочь очистить агрегаты белка, участвующие в нейродегенерации. Оба эти варианта включают в себя очень сложные пути, которые мы только начинаем понимать. [8]
Целью иммунотерапии является улучшение аспектов иммунной системы. От болезни Альцгеймера и других состояний предложены как активная, так и пассивная вакцинация; однако необходимо провести дополнительные исследования, чтобы доказать безопасность и эффективность препарата на людях. [77]
В настоящее время терапевтической мишенью для лечения болезни Альцгеймера является протеаза β-секретаза. [78] [ нужен неосновной источник ] , который участвует в пути амилоидогенной обработки, который приводит к патологическому накоплению белков в мозге. Когда ген, кодирующий белок-предшественник амилоида (APP), подвергается сплайсингу с помощью α-секретазы. [79] [ нужен неосновной источник ] вместо β-секретазы не вырабатывается токсичный белок β-амилоид. Целевое ингибирование [80] β-секретазы потенциально может предотвратить гибель нейронов, ответственную за симптомы болезни Альцгеймера.
Доктор Антонио Барбера, бывший врач акушерства и гинекологии , прописывает настольный теннис пациентам, страдающим серьезным неврологическим расстройством . [81]
См. также
[ редактировать ]Ссылки
[ редактировать ]- ^ Jump up to: а б Эрккинен, Майкл Г.; Ким, Ми-Ок; Гешвинд, Майкл Д. (апрель 2018 г.). «Клиническая неврология и эпидемиология основных нейродегенеративных заболеваний» . Перспективы Колд-Спринг-Харбор в биологии . 10 (4): 20. doi : 10.1101/cshperspect.a033118 . ПМК 5880171 . ПМИД 28716886 .
- ^ Лэмпти Р.Н., Чаулагайн Б., Триведи Р., Готвал А., Лайек Б., Сингх Дж. (февраль 2022 г.). «Обзор распространенных нейродегенеративных заболеваний: современные терапевтические подходы и потенциальная роль нанотерапии» . Int J Mol Sci . 23 (3): 1851. doi : 10.3390/ijms23031851 . ПМЦ 8837071 . ПМИД 35163773 .
- ^ «Основы мозга: жизнь и смерть нейрона | Национальный институт неврологических расстройств и инсульта» . www.ninds.nih.gov . Проверено 12 июля 2024 г.
- ^ Перейра, TMC; Коко, LZ; Тон, АММ; Мейрель, СС; Кампос-Тоймиль, М; Кампаньяро, BP; Васкес, ЕС (20 ноября 2021 г.). «Новый сценарий оси кишечник-мозг: терапевтические действия нового действующего вещества кефира против нейродегенеративных заболеваний» . Антиоксиданты . 10 (11): 1845. doi : 10.3390/antiox10111845 . ПМЦ 8614795 . ПМИД 34829716 .
- ^ Стивенсон, Дж; Нутма, Э; ван дер Валк, П; Амор, С. (июнь 2018 г.). «Воспаление при нейродегенеративных заболеваниях ЦНС» . Иммунология . 154 (2): 204–219. дои : 10.1111/imm.12922 . ПМК 5980185 . ПМИД 29513402 .
- ^ Сингх, А; Кукрети, Р; Сасо, Л; Кукрети, С (22 апреля 2019 г.). «Окислительный стресс: ключевой модулятор нейродегенеративных заболеваний» . Молекулы . 24 (8): 1583. doi : 10.3390/molecules24081583 . ПМК 6514564 . ПМИД 31013638 .
- ^ «Что такое нейродегенеративное заболевание?» . Исследования JPND . 17 июля 2014 года . Проверено 7 февраля 2015 г.
- ^ Jump up to: а б с д и ж г час я Рубинштейн, округ Колумбия (октябрь 2006 г.). «Роль путей внутриклеточной деградации белков в нейродегенерации». Природа . 443 (7113): 780–6. Бибкод : 2006Natur.443..780R . дои : 10.1038/nature05291 . ПМИД 17051204 . S2CID 4411895 .
- ^ Jump up to: а б с д и ж г Бредесен Д.Е., Рао Р.В., Мелен П. (октябрь 2006 г.). «Гибель клеток нервной системы» . Природа . 443 (7113): 796–802. Бибкод : 2006Natur.443..796B . дои : 10.1038/nature05293 . ПМК 3970704 . ПМИД 17051206 .
- ^ Глобальный отчет о мерах общественного здравоохранения по борьбе с деменцией (PDF) . Женева: Всемирная организация здравоохранения. 2021. ISBN 978-92-4-003324-5 . Проверено 14 октября 2022 г.
- ^ Овади, Юдит; Орос, Ференц (2009). Сворачивание и неправильное сворачивание белков: нейродегенеративные заболевания . Сосредоточьтесь на структурной биологии. Дордрехт, Нидерланды, Нью-Йорк, Лондон: Springer. ISBN 978-1-4020-9434-7 .
- ^ «Главная страница журнала» . Клеточная биохимия и биофизика . Спрингер. дои : 10.1007/12013.1559-0283 . ISSN 1559-0283 .
- ^ Венк Г.Л. (2003). «Нейропатологические изменения при болезни Альцгеймера». Журнал клинической психиатрии . 64 (Приложение 9): 7–10. ПМИД 12934968 .
- ^ Замок ММ (27 октября 2013 г.). Загадка Альцгеймера: переплетение деменции и старения . Издательство Принстонского университета. ISBN 978-1-4008-4846-1 . OCLC 859536969 .
- ^ Своб Страц, Дубравка; Конджевод, Марсела; Сагуд, Марина; Николац Перкович, Матеа; Недич Эрьявец, Гордана; Вуик, Барбара; Симич, Горан; Вукич, Вана; Мимика, Нинослав; Пивац, Нела (2021). «Персонализация ухода и лечения болезни Альцгеймера: обзор» . Фармакогеномика и персонализированная медицина . 14 : 631–653. дои : 10.2147/PGPM.S284615 . ПМК 8169052 . ПМИД 34093032 .
- ^ Jump up to: а б Арчер MC, Холл PH, Морган Дж. К. (2017). «[P2–430]: Точность клинической диагностики болезни Альцгеймера в центрах болезни Альцгеймера (ADCS)» . Болезнь Альцгеймера и деменция . 13 (7S_Part_16): 800–1. дои : 10.1016/j.jalz.2017.06.1086 . S2CID 54359937 .
- ^ Приллер С., Бауэр Т., Миттериггер Г., Кребс Б., Кречмар Х.А., Хермс Дж. (июль 2006 г.). «Формирование и функция синапсов модулируются белком-предшественником амилоида» . Журнал неврологии . 26 (27): 7212–21. doi : 10.1523/JNEUROSCI.1450-06.2006 . ПМК 6673945 . ПМИД 16822978 .
- ^ Тернер П.Р., О'Коннор К., Тейт В.П., Авраам В.К. (май 2003 г.). «Роль белка-предшественника амилоида и его фрагментов в регуляции нервной активности, пластичности и памяти». Прогресс нейробиологии . 70 (1): 1–32. дои : 10.1016/S0301-0082(03)00089-3 . ПМИД 12927332 . S2CID 25376584 .
- ^ Хупер, Н.М. (апрель 2005 г.). «Роль протеолиза и липидных рафтов в обработке белка-предшественника амилоида и прионного белка». Труды Биохимического общества . 33 (Часть 2): 335–8. дои : 10.1042/BST0330335 . ПМИД 15787600 .
- ^ Тирабоски П., Хансен Л.А., Тал Л.Дж., Кори-Блум Дж. (июнь 2004 г.). «Важность нейритных бляшек и клубков для развития и эволюции БА». Неврология . 62 (11): 1984–9. дои : 10.1212/01.WNL.0000129697.01779.0A . ПМИД 15184601 . S2CID 25017332 .
- ^ Ониши С., Такано К. (март 2004 г.). «Амилоидные фибриллы с точки зрения сворачивания белка» . Клеточные и молекулярные науки о жизни . 61 (5): 511–524. дои : 10.1007/s00018-003-3264-8 . ПМЦ 11138910 . ПМИД 15004691 . S2CID 25739126 .
- ^ Эльбаз А., Каркайон Л., Каб С., Мойсан Ф. (январь 2016 г.). «Эпидемиология болезни Паркинсона». Неврологический обзор . 172 (1): 14–26. дои : 10.1016/j.neurol.2015.09.012 . ПМИД 26718594 .
- ^ «Обнаружен механизм болезни Паркинсона», HHMI Research News, 22 июня 2006 г.
- ^ Jump up to: а б с Де Вос К.Дж., Грирсон А.Дж., Акерли С., Миллер CC (2008). «Роль аксонального транспорта при нейродегенеративных заболеваниях». Ежегодный обзор неврологии . 31 : 151–73. дои : 10.1146/annurev.neuro.31.061307.090711 . ПМИД 18558852 .
- ^ Jump up to: а б с Варки Дж., Исас Дж.М., Мизуно Н., Дженсен М.Б., Бхатия В.К., Джао CC и др. (октябрь 2010 г.). «Индукция кривизны мембраны и тубуляция являются общими чертами синуклеинов и аполипопротеинов» . Журнал биологической химии . 285 (42): 32486–93. дои : 10.1074/jbc.M110.139576 . ПМЦ 2952250 . ПМИД 20693280 .
- ^ Дэвис А.А., Андруска К.М., Бенитес Б.А., Расетт Б.А., Перлмуттер Дж.С., Кручага К. (январь 2016 г.). «Варианты GBA, SNCA и MAPT влияют на риск болезни Паркинсона, возраст начала и прогрессирование» . Нейробиология старения . 37 : 209.e1–209.e7. doi : 10.1016/j.neurobiolaging.2015.09.014 . ПМК 4688052 . ПМИД 26601739 .
- ^ Jump up to: а б с Шмидт, Неле; Пашен, Лаура; Витт, Карстен (16 ноября 2020 г.). Мирабелла, Джованни (ред.). «Неверная самооценка обонятельного функционирования у пациентов с болезнью Паркинсона может ввести в заблуждение невролога» . Болезнь Паркинсона . 2020 : 1–5. дои : 10.1155/2020/7548394 . ПМЦ 7683170 . ПМИД 33274040 .
- ^ Хилл-Бернс, Эрин М.; Дебелиус, Жюстин В.; Мортон, Джеймс Т.; Виссеманн, Уильям Т.; Льюис, Мэтью Р.; Уоллен, Закари Д.; Педдада, Шьямал Д.; Фактор, Стюарт А.; Молхо, Эрик; Забетян, Сайрус П.; Найт, Роб (май 2017 г.). «Болезнь Паркинсона и лекарства от болезни Паркинсона имеют разные характеристики кишечного микробиома: БП, лекарства и кишечный микробиом» . Двигательные расстройства . 32 (5): 739–749. дои : 10.1002/mds.26942 . ПМК 5469442 . ПМИД 28195358 .
- ^ Первес Д., Августин Г.А., Фитцпатрик Д., Холл В., ЛаМантия А.С., Макнамара Дж.О., Уильямс С.М. (2001). «Цепи в системе базальных ганглиев» . В Первес Д. (ред.). Нейронаука (2-е изд.). Сандерленд, Массачусетс: Sinauer Associates. ISBN 978-0-87893-742-4 . НБК10847.
- ^ Эстрада Санчес А.М., Мехиа-Тойбер Х., Массиу Л. (апрель 2008 г.). «Экситотоксическая гибель нейронов и патогенез болезни Хантингтона». Архивы медицинских исследований . 39 (3): 265–76. doi : 10.1016/j.arcmed.2007.11.011 . ПМИД 18279698 .
- ^ Лобсигер CS, Кливленд DW (ноябрь 2007 г.). «Глиальные клетки как внутренние компоненты неклеточных автономных нейродегенеративных заболеваний» . Природная неврология . 10 (11): 1355–60. дои : 10.1038/nn1988 . ПМК 3110080 . ПМИД 17965655 .
- ^ Purves 2001 , Вставка А. Болезнь Хантингтона
- ^ Кроссман А.Р. (май 2000 г.). «Функциональная анатомия двигательных нарушений» . Журнал анатомии . 196 (Часть 4) (4): 519–25. дои : 10.1046/j.1469-7580.2000.19640519.x . ПМК 1468094 . ПМИД 10923984 .
- ^ Jump up to: а б Барнат, Моня; Капицци, Мариакристина; Апарисио, Эстер; Болуда, Сусана; Веннагель, Дорис; Качер, Радия; Кассем, Райан; Ленуар, Софи; Агасс, Фабьен; Браз, Барбара Ю.; Лю, Дже-Пин (14 августа 2020 г.). «Болезнь Хантингтона изменяет развитие нервной системы человека» . Наука . 369 (6505): 787–793. Бибкод : 2020Sci...369..787B . дои : 10.1126/science.aax3338 . ПМЦ 7859879 . PMID 32675289 .
- ^ Лаббадиа Дж., Моримото Р.И. (август 2013 г.). «Болезнь Хантингтона: основные молекулярные механизмы и новые концепции» . Тенденции биохимических наук . 38 (8): 378–85. дои : 10.1016/j.tibs.2013.05.003 . ПМЦ 3955166 . ПМИД 23768628 .
- ^ Jump up to: а б «Рассеянный склероз: надежда через исследования | Национальный институт неврологических расстройств и инсульта» . www.ninds.nih.gov . Проверено 30 ноября 2020 г.
- ^ Кауфман, Дэвид Майланд; Мильштейн, Марк Дж. (1 января 2013 г.), Кауфман, Дэвид Майланд; Мильштейн, Марк Дж. (ред.), «Глава 15 - Рассеянный склероз» , Клиническая неврология Кауфмана для психиатров (седьмое издание) , Филадельфия: WB Saunders, стр. 329–349, ISBN 978-0-7234-3748-2 , получено 7 декабря 2020 г.
- ^ Jump up to: а б с Стис, Питер К.; Цуцуи, Сигэки (13 декабря 2019 г.). «Последние достижения в понимании рассеянного склероза» . F1000Исследования . 8 : 2100. doi : 10.12688/f1000research.20906.1 . ПМЦ 6915812 . ПМИД 31885862 .
- ^ Ирвин, Калифорния; Блейкмор, ВФ (2008). «Ремиелинизация защищает аксоны от дегенерации аксонов, связанной с демиелинизацией» . Мозг . 131 (6): 1464–77. CiteSeerX 10.1.1.328.2931 . дои : 10.1093/brain/awn080 . ПМИД 18490361 .
- ^ Jump up to: а б с Мин Ю.Г., Чой С.Дж., Хон Ю.Х., Ким СМ, Шин Дж.Ю., Сон Дж.Дж. (сентябрь 2020 г.). «Диссоциированная атрофия мышц ног при боковом амиотрофическом склерозе/заболевании двигательных нейронов: признак «расщепленной ноги»» . Научные отчеты . 10 (1): 15661. Бибкод : 2020НатСР..1015661М . дои : 10.1038/s41598-020-72887-7 . ПМЦ 7518279 . ПМИД 32973334 .
- ^ Jump up to: а б с д Куинн С., Эдмундсон С., Даходвала Н., Элман Л. (апрель 2020 г.). «Надежная и эффективная шкала для оценки бремени заболеваний верхних мотонейронов при боковом амиотрофическом склерозе». Мышцы и нервы . 61 (4): 508–511. дои : 10.1002/mus.26764 . ПМИД 31743477 . S2CID 208186566 .
- ^ Нагай М., Ре Д.Б., Нагата Т., Халазонит А., Джесселл Т.М., Вихтерле Х., Пшедборски С. (май 2007 г.). «Астроциты, экспрессирующие связанные с ALS мутированные факторы высвобождения SOD1, избирательно токсичны для мотонейронов» . Природная неврология . 10 (5): 615–22. дои : 10.1038/nn1876 . ПМЦ 3799799 . ПМИД 17435755 .
- ^ Ди Джорджио Ф.П., Карраско М.А., Сиао М.К., Маниатис Т., Эгган К. (май 2007 г.). «Неклеточное автономное влияние глии на мотонейроны в модели БАС на основе эмбриональных стволовых клеток» . Природная неврология . 10 (5): 608–14. дои : 10.1038/nn1885 . ПМК 3139463 . ПМИД 17435754 .
- ^ Жюльен Дж. П. (май 2007 г.). «АЛС: астроциты становятся смертельными соседями» . Природная неврология . 10 (5): 535–7. дои : 10.1038/nn0507-535 . ПМИД 17453052 . S2CID 2987257 .
- ^ Jump up to: а б «Информационный бюллетень о болезни Баттена | Национальный институт неврологических расстройств и инсульта» . www.ninds.nih.gov . Проверено 30 ноября 2020 г. .
- ^ Jump up to: а б с д и ж Джонсон, Тайлер Б.; Каин, Джейкоб Т.; Уайт, Кэтрин А.; Рамирес-Монтеалегре, Дения; Пирс, Дэвид А.; Веймер, Джилл М. (март 2019 г.). «Терапевтический ландшафт болезни Баттена: современные методы лечения и перспективы на будущее» . Обзоры природы Неврология . 15 (3): 161–178. дои : 10.1038/s41582-019-0138-8 . ПМК 6681450 . ПМИД 30783219 .
- ^ Jump up to: а б с д Мастен М.С., Уильямс Дж.Д., Вермилион Дж., Адамс Х.Р., Вирхил А., Коллинз А. и др. (июнь 2020 г.). «Система стадирования заболевания CLN3: новый инструмент для клинических исследований болезни Баттена» . Неврология . 94 (23): e2436–e2440. дои : 10.1212/WNL.0000000000009454 . ПМЦ 7455368 . ПМИД 32300063 .
- ^ Хартнетт Л. (30 сентября 2019 г.). «Баттенская болезнь». Практика обучения инвалидов . 22 (5): 22. doi : 10.7748/ldp.22.5.22.s16 . S2CID 241832253 .
- ^ «Информационный бюллетень о болезни Крейцфельдта-Якоба | Национальный институт неврологических расстройств и инсульта» . www.ninds.nih.gov . Национальный институт здоровья . Проверено 31 марта 2022 г.
- ^ «Болезнь Крейцфельдта-Якоба – Симптомы и причины» . Клиника Мэйо . Проверено 31 марта 2022 г.
- ^ Исследования, Центр оценки биологических препаратов (12 апреля 2019 г.). «Вариант болезни Крейтцфельдта-Якоба (vCJD) и фактора VIII (pdFVIII), вопросы и ответы» . FDA . Проверено 31 марта 2022 г.
- ^ Jump up to: а б Лин М.Т., Бил М.Ф. (октябрь 2006 г.). «Митохондриальная дисфункция и окислительный стресс при нейродегенеративных заболеваниях». Природа . 443 (7113): 787–95. Бибкод : 2006Natur.443..787L . дои : 10.1038/nature05292 . ПМИД 17051205 . S2CID 4421515 .
- ^ Бернштейн С., Бернштейн Х. (1991). Старение, пол и восстановление ДНК . Сан-Диего: Академическая пресса. стр. 121–139. ISBN 978-0-12-092860-6 .
- ^ Мейнард С., Фанг Э.Ф., Шайби-Кнудсен М., Крото Д.Л., Бор В.А. (сентябрь 2015 г.). «Повреждение ДНК, восстановление ДНК, старение и нейродегенерация» . Перспективы Колд-Спринг-Харбора в медицине . 5 (10): а025130. doi : 10.1101/cshperspect.a025130 . ПМЦ 4588127 . ПМИД 26385091 .
- ^ Камандола С., член парламента от Мэттсона (июнь 2017 г.). «Мозговой метаболизм в здоровье, старении и нейродегенерации» . Журнал ЭМБО . 36 (11): 1474–92. дои : 10.15252/embj.201695810 . ПМК 5452017 . ПМИД 28438892 .
- ^ Jump up to: а б Левин, Кристин С.; Леонард, Хэмптон Л.; Блаувендраат, Корнелис; Иваки, Хиротака; Джонсон, Николас; Бандрес-Сига, Сара; Ферруччи, Луиджи; Фагри, Фараз; Синглтон, Эндрю Б.; Ноллс, Майк А. (19 января 2023 г.). «Воздействие вируса и риск нейродегенеративных заболеваний в национальных биобанках» . Нейрон . 111 (7): 1086–93.e2. дои : 10.1016/j.neuron.2022.12.029 . ПМЦ 10079561 . ПМИД 36669485 .
- Новостная статья об исследовании: Козлов, Макс (23 января 2023 г.). «Масштабный анализ медицинских записей связывает вирусные заболевания с заболеваниями головного мозга» . Природа . 614 (7946): 18–19. Бибкод : 2023Natur.614...18K . дои : 10.1038/d41586-023-00181-3 . ПМИД 36690772 . S2CID 256193462 . Архивировано из оригинала 6 февраля 2023 года . Проверено 15 февраля 2023 г.
- ^ Леблан, Паскаль; Форберг, Ина Майя (4 августа 2022 г.). «Вирусы при нейродегенеративных заболеваниях: больше, чем просто подозреваемые в преступлениях» . ПЛОС Патогены . 18 (8): e1010670. дои : 10.1371/journal.ppat.1010670 . ПМЦ 9352104 . ПМИД 35925897 .
- ^ Jump up to: а б Томпсон Л.М. (апрель 2008 г.). «Нейродегенерация: вопрос баланса» . Природа . 452 (7188): 707–8. Бибкод : 2008Natur.452..707T . дои : 10.1038/452707a . ПМИД 18401401 . S2CID 205037169 .
- ^ Jump up to: а б с Марш Дж.Л., Лукачович Т., Томпсон Л.М. (март 2009 г.). «Животные модели полиглутаминовых заболеваний и терапевтические подходы» . Журнал биологической химии . 284 (12): 7431–5. дои : 10.1074/jbc.R800065200 . ПМК 2658038 . ПМИД 18957429 .
- ^ Орр HT (март 2009 г.). «Серия мини-обзоров нестабильных нуклеотидных повторов: молекулярная биография нарушений нестабильных повторов» . Журнал биологической химии . 284 (12): 7405. doi : 10.1074/jbc.R800067200 . ПМЦ 2658033 . ПМИД 18957428 .
- ^ Зогби Х.И., Орр Х.Т. (март 2009 г.). «Патогенетические механизмы полиглутамин-опосредованного нейродегенеративного заболевания, спиноцеребеллярной атаксии 1 типа» . Журнал биологической химии . 284 (12): 7425–9. дои : 10.1074/jbc.R800041200 . ПМЦ 2658037 . ПМИД 18957430 .
- ^ Набайс, Марта Ф.; Лоус, Саймон М.; Линь, Тиан; Валлерга, Костанца Л.; Армстронг, Никола Дж.; Блэр, Ян П.; Квок, Джон Б.; Мэзер, Карен А.; Меллик, Джордж Д.; Сачдев, Перминдер С.; Уоллес, Линн (26 марта 2021 г.). «Метаанализ полногеномного метилирования ДНК выявляет общие ассоциации при нейродегенеративных расстройствах» . Геномная биология . 22 (1): 90. дои : 10.1186/s13059-021-02275-5 . ПМК 8004462 . ПМИД 33771206 .
- ^ Чанг, CG; Ли, Х; Ли, SB (сентябрь 2018 г.). «Механизмы белковой токсичности при нейродегенеративных заболеваниях» . Клеточные и молекулярные науки о жизни . 75 (17): 3159–80. дои : 10.1007/s00018-018-2854-4 . ПМК 6063327 . ПМИД 29947927 .
- ^ ДиМауро С., Шон Э.А. (2008). «Митохондриальные нарушения в нервной системе». Ежегодный обзор неврологии . 31 : 91–123. дои : 10.1146/annurev.neuro.30.051606.094302 . ПМИД 18333761 .
- ^ Лю З, Чжоу Т, Циглер А.С., Димитрион П., Цзо Л. (2017). «Окислительный стресс при нейродегенеративных заболеваниях: от молекулярных механизмов к клиническому применению» . Окислительная медицина и клеточное долголетие . 2017 : 2525967. дои : 10.1155/2017/2525967 . ПМК 5529664 . ПМИД 28785371 .
- ^ Jump up to: а б Ван Х., Дхармалингам П., Васкес В., Митра Дж., Болдог И., Рао К.С. и др. (январь 2017 г.). «Хроническое окислительное повреждение вместе с дефицитом восстановления генома в нейронах является двойным ударом для нейродегенерации: сигнализирует ли реакция повреждения о потенциальной терапевтической цели?» . Механизмы старения и развития . 161 (Часть А): 163–176. дои : 10.1016/j.mad.2016.09.005 . ПМК 5316312 . ПМИД 27663141 .
- ^ Jump up to: а б Мадабхуши Р., Пан Л., Цай Л.Х. (июль 2014 г.). «Повреждение ДНК и его связь с нейродегенерацией» . Нейрон . 83 (2): 266–282. дои : 10.1016/j.neuron.2014.06.034 . ПМК 5564444 . ПМИД 25033177 .
- ^ Jump up to: а б с Йеппесен Д.К., Бор В.А., Стевнснер Т. (июль 2011 г.). «Дефицит репарации ДНК при нейродегенерации» . Прогресс нейробиологии . 94 (2): 166–200. doi : 10.1016/j.pneurobio.2011.04.013 . ПМЦ 3123739 . ПМИД 21550379 .
- ^ Коулман, член парламента, Фриман, М.Р. (2010). «Валлеровская дегенерация, wld(s) и nmnat» . Анну преподобный Neurosci . 33 : 245–67. doi : 10.1146/annurev-neuro-060909-153248 . ПМЦ 5223592 . ПМИД 20345246 .
- ^ Энгельберг-Кулка Х., Амитай С., Колодкин-Гал И., Хазан Р. (октябрь 2006 г.). «Бактериальная запрограммированная гибель клеток и многоклеточное поведение бактерий» . ПЛОС Генетика . 2 (10): е135. дои : 10.1371/journal.pgen.0020135 . ПМК 1626106 . ПМИД 17069462 .
- ^ Вила М, Пшедборски С (май 2003 г.). «Нацеливание на запрограммированную гибель клеток при нейродегенеративных заболеваниях». Обзоры природы. Нейронаука . 4 (5): 365–75. дои : 10.1038/nrn1100 . ПМИД 12728264 . S2CID 33018251 .
- ^ Грин Д.Р., Кремер Г. (октябрь 2005 г.). «Фармакологическое манипулирование гибелью клеток: скоро клиническое применение?» . Журнал клинических исследований . 115 (10): 2610–7. дои : 10.1172/JCI26321 . ПМЦ 1236695 . ПМИД 16200193 .
- ^ Jump up to: а б с д и ж г час я Каккамо Д., Курро М., Конделло С., Ферлаццо Н., Иентиле Р. (февраль 2010 г.). «Критическая роль трансглутаминазы и других белков стресса во время нейродегенеративных процессов». Аминокислоты . 38 (2): 653–8. дои : 10.1007/s00726-009-0428-3 . ПМИД 19960212 . S2CID 19739739 .
- ^ Димебон разочаровывается в испытании фазы 3
- ^ Свитлав М (2012). «Фаза III КОНЦЕРТА. Испытание латрепирдина. Отрицательные результаты». Фарм Мед . 26 (2): 113–5. дои : 10.1007/BF03256900 .
- ^ Галоян А.А., Саркисян Д.С., Чавушян В.А., Меликсетян И.Б., Авагян З.Е., Погосян М.В. и др. (сентябрь 2008 г.). «Нейропротекция гипоталамического пептида, богатого пролином, пептида-1 в модели болезни Альцгеймера Abeta25-35». Болезнь Альцгеймера и деменция . 4 (5): 332–44. дои : 10.1016/j.jalz.2007.10.019 . ПМИД 18790460 . S2CID 39817779 .
- ^ Броуди Д.Л., Хольцман Д.М. (2008). «Активная и пассивная иммунотерапия нейродегенеративных заболеваний» . Ежегодный обзор неврологии . 31 : 175–93. дои : 10.1146/annurev.neuro.31.060407.125529 . ПМК 2561172 . ПМИД 18352830 .
- ^ Пасторино Л., Икин А.Ф., Ламприану С., Вакарес Н., Ревелли Дж.П., Платт К. и др. (апрель 2004 г.). «BACE (бета-секретаза) модулирует обработку APLP2 in vivo». Молекулярная и клеточная нейронауки . 25 (4): 642–9. дои : 10.1016/j.mcn.2003.12.013 . ПМИД 15080893 . S2CID 54334969 .
- ^ Эш Ф.С., Кейм П.С., Битти ЕС, Блахер Р.В., Калвелл А.Р., Олтерсдорф Т. и др. (июнь 1990 г.). «Расщепление бета-амилоидного пептида во время конститутивного процессинга его предшественника». Наука . 248 (4959): 1122–4. Бибкод : 1990Sci...248.1122E . дои : 10.1126/science.2111583 . ПМИД 2111583 .
- ^ Шенк Д., Баси Г.С., Пангалос М.Н. (сентябрь 2012 г.). «Стратегии лечения, направленные на β-амилоидный белок» . Перспективы Колд-Спринг-Харбора в медицине . 2 (9): а006387. doi : 10.1101/cshperspect.a006387 . ПМЦ 3426815 . ПМИД 22951439 .
- ^ «Врач из Колорадо прописывает лечение нейродегенеративных заболеваний пинг-понгом: «Делать что-нибудь хорошее» » . Фокс Ньюс . 12 января 2024 г.