болезнь Хантингтона
| болезнь Хантингтона | |
|---|---|
| Другие имена | хорея Гентингтона |
 | |
| Отредактированное микроскопическое изображение среднего шиповидного нейрона (желтый) с телом включения (оранжевый), которое возникает как часть болезненного процесса (ширина изображения 360 мкм ). | |
| Specialty | Neurology |
| Symptoms | Problems with motor skills, including coordination and gait, mood, and mental abilities[1][2] |
| Complications | Pneumonia, heart disease, physical injury from falls, suicide[3] |
| Usual onset | 30–50 years old[4] |
| Duration | Long term[4] |
| Causes | Genetic (inherited or new mutation)[4] |
| Diagnostic method | Genetic testing[5] |
| Differential diagnosis | Sydenham's chorea, benign hereditary chorea, lupus, paraneoplastic syndrome, Wilson's disease[6] |
| Treatment | Supportive care[2] |
| Medication | Tetrabenazine[3] |
| Prognosis | 15–20 years from onset of symptoms[4] |
| Frequency | 4–15 in 100,000 (European descent)[1] |
| Named after | George Huntington |
Болезнь Хантингтона ( БХ ), также известная как хорея Хантингтона , является неизлечимым нейродегенеративным заболеванием. [ 7 ] это в основном наследуется . [ 8 ] Самыми ранними симптомами часто являются едва заметные проблемы с настроением или умственными/психиатрическими способностями. [ 9 ] [ 1 ] Часто за этим следует общее отсутствие координации и неустойчивая походка . [ 2 ] Это также заболевание базальных ганглиев, вызывающее гиперкинетическое двигательное расстройство , известное как хорея . [ 10 ] [ 11 ] По мере прогрессирования заболевания нескоординированные, непроизвольные движения тела при хорее становятся более очевидными. [ 1 ] Физические способности постепенно ухудшаются, пока координация движений не становится затрудненной и человек не может говорить. [ 1 ] [ 2 ] Умственные способности обычно снижаются до деменции , депрессии, апатии и временами импульсивности. [ 9 ] [ 12 ] [ 3 ] Конкретные симптомы несколько различаются у разных людей. [ 1 ] Симптомы обычно начинаются в возрасте от 30 до 50 лет и могут начаться в любом возрасте, но обычно наблюдаются в возрасте около 40 лет. [ 12 ] [ 9 ] [ 3 ] [ 4 ] Заболевание может развиваться раньше в каждом последующем поколении . [ 1 ] Около восьми процентов случаев начинаются в возрасте до 20 лет и известны как ювенильная ГБ , которая обычно проявляется медленными симптомами болезни Паркинсона , а не хореи. [ 3 ]
HD is typically inherited from an affected parent, who carries a mutation in the huntingtin gene (HTT).[4] However, up to 10% of cases are due to a new mutation.[1] The huntingtin gene provides the genetic information for huntingtin protein (Htt).[1] Expansion of CAG repeats of cytosine-adenine-guanine (known as a trinucleotide repeat expansion) in the gene coding for the huntingtin protein results in an abnormal mutant protein (mHtt), which gradually damages brain cells through a number of possible mechanisms.[8][13] The mutant protein is dominant, so having one parent who is a carrier of the trait is sufficient to trigger the disease in their children. Diagnosis is by genetic testing, which can be carried out at any time, regardless of whether or not symptoms are present.[5] This fact raises several ethical debates: the age at which an individual is considered mature enough to choose testing; whether parents have the right to have their children tested; and managing confidentiality and disclosure of test results.[2]
No cure for HD is known, and full-time care is required in the later stages.[2] Treatments can relieve some symptoms and in some, improve quality of life.[3] The best evidence for treatment of the movement problems is with tetrabenazine.[3] HD affects about 4 to 15 in 100,000 people of European descent.[1][3] It is rare among the Finnish and Japanese, while the occurrence rate in Africa is unknown.[3] The disease affects males and females equally.[3] Complications such as pneumonia, heart disease, and physical injury from falls reduce life expectancy; although fatal aspiration pneumonia is commonly cited as the ultimate cause of death for those with the condition.[14][12][3] Suicide is the cause of death in about 9% of cases.[3] Death typically occurs 15–20 years from when the disease was first detected.[4]
The earliest known description of the disease was in 1841 by American physician Charles Oscar Waters.[15] The condition was described in further detail in 1872 by American physician George Huntington.[15] The genetic basis was discovered in 1993 by an international collaborative effort led by the Hereditary Disease Foundation.[16][17] Research and support organizations began forming in the late 1960s to increase public awareness, provide support for individuals and their families and promote research.[17][18] Research directions include determining the exact mechanism of the disease, improving animal models to aid with research, testing of medications and their delivery to treat symptoms or slow the progression of the disease, and studying procedures such as stem-cell therapy with the goal of replacing damaged or lost neurons.[16]
Signs and symptoms
[edit]Signs and symptoms of Huntington's disease most commonly become noticeable between the ages of 30 and 50 years, but they can begin at any age[4] and present as a triad of motor, cognitive, and psychiatric symptoms.[19] When developed in an early stage, it is known as juvenile Huntington's disease.[20] In 50% of cases, the psychiatric symptoms appear first.[19] Their progression is often described in early stages, middle stages, and late stages with an earlier prodromal phase.[2] In the early stages, subtle personality changes, problems in cognition and physical skills, irritability, and mood swings occur, all of which may go unnoticed,[21][22] and these usually precede the motor symptoms.[23] Almost everyone with HD eventually exhibits similar physical symptoms, but the onset, progression, and extent of cognitive and behavioral symptoms vary significantly between individuals.[24][25]
The most characteristic initial physical symptoms are jerky, random, and uncontrollable movements called chorea.[10] Many people are not aware of their involuntary movements, or impeded by them.[1] Chorea may be initially exhibited as general restlessness, small unintentionally initiated or uncompleted motions, lack of coordination, or slowed saccadic eye movements.[26] These minor motor abnormalities usually precede more obvious signs of motor dysfunction by at least three years.[27] The clear appearance of symptoms such as rigidity, writhing motions, or abnormal posturing appear as the disorder progresses.[26] These are signs that the system in the brain that is responsible for movement has been affected.[28] Psychomotor functions become increasingly impaired, such that any action that requires muscle control is affected. When muscle control is affected such as rigidity or muscle contracture this is known as dystonia. Dystonia is a neurological hyperkinetic movement disorder that results in twisting or repetitive movements, that may resemble a tremor. Common consequences are physical instability, abnormal facial expression, and difficulties chewing, swallowing, and speaking.[26] Sleep disturbances and weight loss are also associated symptoms.[29] Eating difficulties commonly cause weight loss and may lead to malnutrition.[30][31] Weight loss is common in people with Huntington's disease, and it progresses with the disease. Juvenile HD generally progresses at a faster rate with greater cognitive decline, and chorea is exhibited briefly, if at all; the Westphal variant of slowness of movement, rigidity, and tremors is more typical in juvenile HD, as are seizures.[26][29]
Cognitive abilities are progressively impaired and tend to generally decline into dementia.[3] Especially affected are executive functions, which include planning, cognitive flexibility, abstract thinking, rule acquisition, initiation of appropriate actions, and inhibition of inappropriate actions. Different cognitive impairments include difficulty focusing on tasks, lack of flexibility, a lack of impulse, a lack of awareness of one's own behaviors and abilities and difficulty learning or processing new information. As the disease progresses, memory deficits tend to appear. Reported impairments range from short-term memory deficits to long-term memory difficulties, including deficits in episodic (memory of one's life), procedural (memory of the body of how to perform an activity), and working memory.[28]
Reported neuropsychiatric signs are anxiety, depression, a reduced display of emotions, egocentrism, aggression, and compulsive behavior and hallucination and delusion.[32] Other common psychiatric disorders could include obsessive–compulsive disorder, mania, insomnia and bipolar disorder. Difficulties in recognizing other people's negative expressions have also been observed.[28] The prevalence of these symptoms is highly variable between studies, with estimated rates for lifetime prevalence of psychiatric disorders between 33 and 76%.[32] For many with the disease and their families, these symptoms are among the most distressing aspects of the disease, often affecting daily functioning and constituting reason for institutionalization.[32] Early behavioral changes in HD result in an increased risk of suicide.[10] Often, individuals have reduced awareness of chorea, cognitive, and emotional impairments.[33]
Mutant huntingtin is expressed throughout the body and associated with abnormalities in peripheral tissues that are directly caused by such expression outside the brain. These abnormalities include muscle atrophy, cardiac failure, impaired glucose tolerance, weight loss, osteoporosis, and testicular atrophy.[34]
Genetics
[edit]Everyone has two copies of the huntingtin gene (HTT), which codes for the huntingtin protein (Htt). HTT is also called the HD gene, and the IT15 gene, (interesting transcript 15). Part of this gene is a repeated section called a trinucleotide repeat expansion – a short repeat, which varies in length between individuals, and may change length between generations. If the repeat is present in a healthy gene, a dynamic mutation may increase the repeat count and result in a defective gene. When the length of this repeated section reaches a certain threshold, it produces an altered form of the protein, called mutant huntingtin protein (mHtt). The differing functions of these proteins are the cause of pathological changes, which in turn cause the disease symptoms. The Huntington's disease mutation is genetically dominant and almost fully penetrant; mutation of either of a person's HTT alleles causes the disease. It is not inherited according to sex, but by the length of the repeated section of the gene; hence its severity can be influenced by the sex of the affected parent.[26]
Genetic mutation
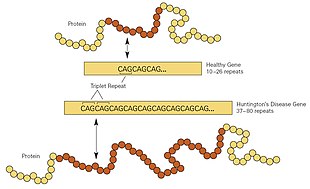
[edit]HD is one of several trinucleotide repeat disorders that are caused by the length of a repeated section of a gene exceeding a normal range.[26] The HTT gene is located on the short arm of chromosome 4[26] at 4p16.3. HTT contains a sequence of three DNA bases—cytosine-adenine-guanine (CAG)—repeated multiple times (i.e. ... CAGCAGCAG ...), known as a trinucleotide repeat.[26] CAG is the three-letter genetic code (codon) for the amino acid glutamine, so a series of them results in the production of a chain of glutamine known as a polyglutamine tract (or polyQ tract), and the repeated part of the gene, the polyQ region.[35]
| Repeat count | Classification | Disease status | Risk to offspring |
|---|---|---|---|
| <27 | Normal | Will not be affected | None |
| 27–35 | Intermediate | Will not be affected | Elevated, but <50% |
| 36–39 | Reduced Penetrance | May or may not be affected | 50% |
| 40+ | Full penetrance | Will be affected | 50% |
Generally, people have fewer than 36 repeated glutamines in the polyQ region, which results in the production of the cytoplasmic protein huntingtin.[26] However, a sequence of 36 or more glutamines results in the production of a protein with different characteristics.[26] This altered form, called mutant huntingtin (mHtt), increases the decay rate of certain types of neurons. Regions of the brain have differing amounts and reliance on these types of neurons and are affected accordingly.[26] Generally, the number of CAG repeats is related to how much this process is affected, and accounts for about 60% of the variation of the age of the onset of symptoms. The remaining variation is attributed to the environment and other genes that modify the mechanism of HD.[26] About 36 to 39 repeats result in a reduced-penetrance form of the disease, with a much later onset and slower progression of symptoms. In some cases, the onset may be so late that symptoms are never noticed.[26] With very large repeat counts (more than 60), HD onset can occur below the age of 20, known as juvenile HD. Juvenile HD is typically of the Westphal variant that is characterized by slowness of movement, rigidity, and tremors. This accounts for about 7% of HD carriers.[36][37]
Inheritance
[edit]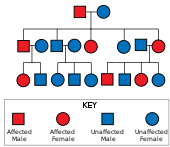
Huntington's disease has autosomal dominant inheritance, meaning that an affected individual typically inherits one copy of the gene with an expanded trinucleotide repeat (the mutant allele) from an affected parent.[26] Since the penetrance of the mutation is very high, those who have a mutated copy of the gene will have the disease. In this type of inheritance pattern, each offspring of an affected individual has a 50% risk of inheriting the mutant allele, so are affected with the disorder (see figure). This probability is sex-independent.[38] Sex-dependent or sex-linked genes are traits that are found on the X or Y chromosomes.[39]
Trinucleotide CAG repeats numbering over 28 are unstable during replication, and this instability increases with the number of repeats present.[26] This usually leads to new expansions as generations pass (dynamic mutations) instead of reproducing an exact copy of the trinucleotide repeat.[26] This causes the number of repeats to change in successive generations, such that an unaffected parent with an "intermediate" number of repeats (28–35), or "reduced penetrance" (36–40), may pass on a copy of the gene with an increase in the number of repeats that produces fully penetrant HD.[26] The earlier age of onset and greater severity of disease in successive generations due to increases in the number of repeats is known as genetic anticipation.[1] Instability is greater in spermatogenesis than oogenesis;[26] maternally inherited alleles are usually of a similar repeat length, whereas paternally inherited ones have a higher chance of increasing in length.[26][40] Rarely is Huntington's disease caused by a new mutation, where neither parent has over 36 CAG repeats.[41]
In the rare situations where both parents have an expanded HD gene, the risk increases to 75%, and when either parent has two expanded copies, the risk is 100% (all children will be affected). Individuals with both genes affected are rare. For some time, HD was thought to be the only disease for which possession of a second mutated gene did not affect symptoms and progression,[42] but it has since been found that it can affect the phenotype and the rate of progression.[26][43]
Mechanisms
[edit]Huntingtin protein interacts with over 100 other proteins, and appears to have multiple functions.[44] The behavior of the mutated protein (mHtt) is not completely understood, but it is toxic to certain cell types, particularly brain cells. Early damage is most evident in the subcortical basal ganglia, initially in the striatum, but as the disease progresses, other areas of the brain are also affected, including regions of the cerebral cortex. Early symptoms are attributable to functions of the striatum and its cortical connections—namely control over movement, mood, and higher cognitive function.[26] DNA methylation also appears to be changed in HD.[45]
Huntingtin function
[edit]Htt is expressed in all cells, with the highest concentrations found in the brain and testes, and moderate amounts in the liver, heart, and lungs. Its functions are unclear, but it does interact with proteins involved in transcription, cell signaling, and intracellular transporting.[46] In animals genetically modified to exhibit HD, several functions of Htt have been identified.[47] In these animals, Htt is important for embryonic development, as its absence is related to embryonic death. Caspase, an enzyme which plays a role in catalyzing apoptosis, is thought to be activated by the mutated gene through damaging the ubiquitin-protease system. It also acts as an antiapoptotic agent preventing programmed cell death and controls the production of brain-derived neurotrophic factor, a protein that protects neurons and regulates their creation during neurogenesis. Htt also facilitates synaptic vesicular transport and synaptic transmission, and controls neuronal gene transcription.[47] If the expression of Htt is increased, brain cell survival is improved and the effects of mHtt are reduced, whereas when the expression of Htt is reduced, the resulting characteristics are more as seen in the presence of mHtt.[47] Accordingly, the disease is thought not to be caused by inadequate production of Htt, but by a toxic gain-of-function of mHtt in the body.[26]
Cellular changes
[edit]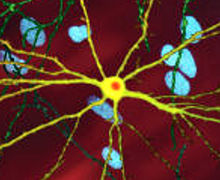
The toxic action of mHtt may manifest and produce the HD pathology through multiple cellular changes.[48][49] In its mutant (polyglutamine expanded) form, the protein is more prone to cleavage that creates shorter fragments containing the polyglutamine expansion.[48] These protein fragments have a propensity to undergo misfolding and aggregation, yielding fibrillar aggregates in which non-native polyglutamine β-strands from multiple proteins are bonded together by hydrogen bonds.[13] These aggregates share the same fundamental cross-beta amyloid architecture seen in other protein deposition diseases .[50] Over time, the aggregates accumulate to form inclusion bodies within cells, ultimately interfering with neuronal function.[13][48] Inclusion bodies have been found in both the cell nucleus and cytoplasm.[48] Inclusion bodies in cells of the brain are one of the earliest pathological changes, and some experiments have found that they can be toxic for the cell, but other experiments have shown that they may form as part of the body's defense mechanism and help protect cells.[48]
Several pathways by which mHtt may cause cell death have been identified. These include effects on chaperone proteins, which help fold proteins and remove misfolded ones; interactions with caspases, which play a role in the process of removing cells; the toxic effects of glutamine on nerve cells; impairment of energy production within cells; and effects on the expression of genes.[13][51]
Mutant huntingtin protein has been found to play a key role in mitochondrial dysfunction.[46] The impairment of mitochondrial electron transport can result in higher levels of oxidative stress and release of reactive oxygen species.[52]
Glutamine is known to be excitotoxic when present in large amounts, that can cause damage to numerous cellular structures. Excessive glutamine is not found in HD, but the interactions of the altered huntingtin protein with numerous proteins in neurons lead to an increased vulnerability to glutamine. The increased vulnerability is thought to result in excitotoxic effects from normal glutamine levels.[13]
Macroscopic changes
[edit]
Initially, damage to the brain is regionally specific with the dorsal striatum in the subcortical basal ganglia being primarily affected, followed later by cortical involvement in all areas.[53][54] Other areas of the basal ganglia affected include the substantia nigra; cortical involvement includes cortical layers 3, 5, and 6; also evident is involvement of the hippocampus, Purkinje cells in the cerebellum, lateral tuberal nuclei of the hypothalamus and parts of the thalamus.[26] These areas are affected according to their structure and the types of neurons they contain, reducing in size as they lose cells.[26] Striatal medium spiny neurons are the most vulnerable, particularly ones with projections towards the external globus pallidus, with interneurons and spiny cells projecting to the internal globus pallidus being less affected.[26][55] HD also causes an abnormal increase in astrocytes and activation of the brain's immune cells, microglia.[56]
The basal ganglia play a key role in movement and behavior control. Their functions are not fully understood, but theories propose that they are part of the cognitive executive system[28] and the motor circuit.[57] The basal ganglia ordinarily inhibit a large number of circuits that generate specific movements. To initiate a particular movement, the cerebral cortex sends a signal to the basal ganglia that causes the inhibition to be released. Damage to the basal ganglia can cause the release or reinstatement of the inhibitions to be erratic and uncontrolled, which results in an awkward start to the motion or motions to be unintentionally initiated or in a motion to be halted before or beyond its intended completion. The accumulating damage to this area causes the characteristic erratic movements associated with HD known as chorea, a dyskinesia.[57] Because of the basal ganglia's inability to inhibit movements, individuals affected by it inevitably experience a reduced ability to produce speech and swallow foods and liquids (dysphagia).[58]
Transcriptional dysregulation
[edit]CREB-binding protein (CBP), a transcriptional coregulator, is essential for cell function because as a coactivator at a significant number of promoters, it activates the transcription of genes for survival pathways.[51] CBP contains an acetyltransferase domain to which HTT binds through its polyglutamine-containing domain.[59] Autopsied brains of those who had Huntington's disease also have been found to have incredibly reduced amounts of CBP.[60] In addition, when CBP is overexpressed, polyglutamine-induced death is diminished, further demonstrating that CBP plays an important role in Huntington's disease and neurons in general.[51]
Diagnosis
[edit]Diagnosis of the onset of HD can be made following the appearance of physical symptoms specific to the disease.[26] Genetic testing can be used to confirm a physical diagnosis if no family history of HD exists. Even before the onset of symptoms, genetic testing can confirm if an individual or embryo carries an expanded copy of the trinucleotide repeat (CAG) in the HTT gene that causes the disease. Genetic counseling is available to provide advice and guidance throughout the testing procedure and on the implications of a confirmed diagnosis. These implications include the impact on an individual's psychology, career, family-planning decisions, relatives, and relationships. Despite the availability of pre-symptomatic testing, only 5% of those at risk of inheriting HD choose to do so.[26]
Clinical
[edit]
A physical examination, sometimes combined with a psychological examination, can determine whether the onset of the disease has begun.[26] Excessive unintentional movements of any part of the body are often the reason for seeking medical consultation. If these are abrupt and have random timing and distribution, they suggest a diagnosis of HD. Cognitive or behavioral symptoms are rarely the first symptoms diagnosed; they are usually only recognized in hindsight or when they develop further. How far the disease has progressed can be measured using the unified Huntington's disease rating scale, which provides an overall rating system based on motor, behavioral, cognitive, and functional assessments.[62][63] Medical imaging, such as a CT scan or MRI scan, can show atrophy of the caudate nuclei early in the disease, as seen in the illustration to the right, but these changes are not, by themselves, diagnostic of HD. Cerebral atrophy can be seen in the advanced stages of the disease. Functional neuroimaging techniques, such as functional magnetic resonance imaging (fMRI) and positron emission tomography (PET), can show changes in brain activity before the onset of physical symptoms, but they are experimental tools and are not used clinically.[26]
Predictive genetic testing
[edit]Because HD follows an autosomal dominant pattern of inheritance, a strong motivation exists for individuals who are at risk of inheriting it to seek a diagnosis. The genetic test for HD consists of a blood test, which counts the numbers of CAG repeats in each of the HTT alleles.[64] Cutoffs are given as follows:
- At 40 or more CAG repeats, full penetrance allele (FPA) exists.[65] A "positive test" or "positive result" generally refers to this case. A positive result is not considered a diagnosis, since it may be obtained decades before the symptoms begin. However, a negative test means that the individual does not carry the expanded copy of the gene and will not develop HD.[26] The test will tell a person who originally had a 50% chance of inheriting the disease if their risk goes up to 100% or is eliminated. Persons who test positive for the disease will develop HD sometime within their lifetimes, provided they live long enough for the disease to appear.[26]
- At 36 to 39 repeats, incomplete or reduced penetrance allele (RPA) may cause symptoms, usually later in the adult life.[65] The maximum risk is 60% that a person with an RPA will be symptomatic at age 65, and 70% at 75.[65]
- At 27 to 35 repeats, intermediate allele (IA), or large normal allele, is not associated with symptomatic disease in the tested individual, but may expand upon further inheritance to give symptoms in offspring.[65]
- With 26 or fewer repeats, the result is not associated with HD.[65]
Testing before the onset of symptoms is a life-changing event and a very personal decision.[26] The main reason given for choosing to test for HD is to aid in career and family decisions.[26] Predictive testing for Huntington's disease has been available via linkage analysis (which requires testing multiple family members) since 1986 and via direct mutation analysis since 1993.[66] At that time, surveys indicated that 50–70% of at-risk individuals would have been interested in receiving testing, but since predictive testing has been offered far fewer choose to be tested.[67] Over 95% of individuals at risk of inheriting HD do not proceed with testing, mostly because it has no treatment.[26] A key issue is the anxiety an individual experiences about not knowing whether they will eventually develop HD, compared to the impact of a positive result.[26] Irrespective of the result, stress levels are lower two years after being tested, but the risk of suicide is increased after a positive test result.[26] Individuals found to have not inherited the disorder may experience survivor guilt about family members who are affected.[26] Other factors taken into account when considering testing include the possibility of discrimination and the implications of a positive result, which usually means a parent has an affected gene and that the individual's siblings will be at risk of inheriting it.[26] In one study, genetic discrimination was found in 46% of individuals at risk for Huntington's disease. It occurred at higher rates within personal relationships than health insurance or employment relations.[68] Genetic counseling in HD can provide information, advice and support for initial decision-making, and then, if chosen, throughout all stages of the testing process.[69] Because of the implications of this test, patients who wish to undergo testing must complete three counseling sessions which provide information about Huntington's.[70]
Counseling and guidelines on the use of genetic testing for HD have become models for other genetic disorders, such as autosomal dominant cerebellar ataxia.[26][71][72] Presymptomatic testing for HD has also influenced testing for other illnesses with genetic variants such as polycystic kidney disease, familial Alzheimer's disease and breast cancer.[71] The European Molecular Genetics Quality Network have published yearly external quality assessment scheme for molecular genetic testing for this disease and have developed best practice guidelines for genetic testing for HD to assist in testing and reporting of results.[73]
Preimplantation genetic diagnosis
[edit]Embryos produced using in vitro fertilization may be genetically tested for HD using preimplantation genetic diagnosis. This technique, where one or two cells are extracted from a typically 4- to 8-cell embryo and then tested for the genetic abnormality, can then be used to ensure embryos affected with HD genes are not implanted, so any offspring will not inherit the disease. Some forms of preimplantation genetic diagnosis—non-disclosure or exclusion testing—allow at-risk people to have HD-free offspring without revealing their own parental genotype, giving no information about whether they themselves are destined to develop HD. In exclusion testing, the embryo's DNA is compared with that of the parents and grandparents to avoid inheritance of the chromosomal region containing the HD gene from the affected grandparent. In nondisclosure testing, only disease-free embryos are replaced in the uterus while the parental genotype and hence parental risk for HD are never disclosed.[74][75]
Prenatal testing
[edit]Obtaining a prenatal diagnosis for an embryo or fetus in the womb is also possible, using fetal genetic material acquired through chorionic villus sampling. An amniocentesis can be performed if the pregnancy is further along, within 14–18 weeks. This procedure looks at the amniotic fluid surrounding the baby for indicators of the HD mutation.[76] This, too, can be paired with exclusion testing to avoid disclosure of parental genotype. Prenatal testing can be done when parents have been diagnosed with HD, when they have had genetic testing showing the expansion of the HTT gene, or when they have a 50% chance of inheriting the disease. The parents can be counseled on their options, which include termination of pregnancy, and on the difficulties of a child with the identified gene.[77][78]
In addition, in at-risk pregnancies due to an affected male partner, noninvasive prenatal diagnosis can be performed by analyzing cell-free fetal DNA in a blood sample taken from the mother (via venipuncture) between six and 12 weeks of pregnancy.[65] It has no procedure-related risk of miscarriage.[65]
Differential diagnosis
[edit]About 99% of HD diagnoses based on the typical symptoms and a family history of the disease are confirmed by genetic testing to have the expanded trinucleotide repeat that causes HD. Most of the remaining are called HD-like (HDL) syndromes.[26][79] The cause of most HDL diseases is unknown, but those with known causes are due to mutations in the prion protein gene (HDL1), the junctophilin 3 gene (HDL2), a recessively inherited unknown gene (HDL3—only found in two families and poorly understood), and the gene encoding the TATA box-binding protein (SCA17, sometimes called HDL4). Other autosomal dominant diseases that can be misdiagnosed as HD are dentatorubral-pallidoluysian atrophy and neuroferritinopathy. Also, some autosomal recessive disorders resemble sporadic cases of HD. These include chorea acanthocytosis and pantothenate kinase-associated neurodegeneration. One X-linked disorder of this type is McLeod syndrome.[79]
Management
[edit]
Treatments are available to reduce the severity of some HD symptoms.[80] For many of these treatments, evidence to confirm their effectiveness in treating symptoms of HD specifically are incomplete.[26][81] As the disease progresses, the ability to care for oneself declines, and carefully managed multidisciplinary caregiving becomes increasingly necessary.[26] Although relatively few studies of exercises and therapies have shown to be helpful to rehabilitate cognitive symptoms of HD, some evidence shows the usefulness of physical therapy, occupational therapy, and speech therapy.[26]
Therapy
[edit]Weight loss and problems in eating due to dysphagia and other muscle discoordination are common, making nutrition management increasingly important as the disease advances.[26] Thickening agents can be added to liquids, as thicker fluids are easier and safer to swallow.[26] Reminding the affected person to eat slowly and to take smaller pieces of food into the mouth may also be of use to prevent choking.[26] If eating becomes too hazardous or uncomfortable, the option of using a percutaneous endoscopic gastrostomy is available. This feeding tube, permanently attached through the abdomen into the stomach, reduces the risk of aspirating food and provides better nutritional management.[82] Assessment and management by speech-language pathologists with experience in Huntington's disease is recommended.[26]
People with Huntington's disease may see a physical therapist for noninvasive and nonmedication-based ways of managing the physical symptoms. Physical therapists may implement fall risk assessment and prevention, as well as strengthening, stretching, and cardiovascular exercises. Walking aids may be prescribed as appropriate. Physical therapists also prescribe breathing exercises and airway clearance techniques with the development of respiratory problems.[83] Consensus guidelines on physiotherapy in Huntington's disease have been produced by the European HD Network.[83] Goals of early rehabilitation interventions are prevention of loss of function. Participation in rehabilitation programs during the early to middle stage of the disease may be beneficial as it translates into long-term maintenance of motor and functional performance. Rehabilitation during the late stage aims to compensate for motor and functional losses.[84] For long-term independent management, the therapist may develop home exercise programs for appropriate people.[85]
Additionally, an increasing number of people with HD are turning to palliative care, which aims to improve quality of life through the treatment of the symptoms and stress of serious illness, in addition to their other treatments.[86]
Medications
[edit]
Tetrabenazine was approved in 2000 for treatment of chorea in Huntington's disease in the EU, and in 2008 in the US.[87] Although other drugs had been used "off label", tetrabenazine was the first approved treatment for Huntington's disease in the U.S. The compound has been known since the 1950s. An alternative to tetrabenazine is amantadine but there is limited evidence for its safety and efficacy.[88]
Other drugs that help to reduce chorea include antipsychotics and benzodiazepines.[22] Hypokinesia and rigidity, especially in juvenile cases, can be treated with antiparkinsonian drugs, and myoclonic hyperkinesia can be treated with valproic acid.[22] Tentative evidence has found ethyl eicosapentaenoic acid to improve motor symptoms at one year.[89] In 2017, deutetrabenazine, a heavier form of tetrabenazine medication for the treatment of chorea in HD, was approved by the FDA.[90] This is marketed as Austedo.
Psychiatric symptoms can be treated with medications similar to those used in the general population.[26][81] Selective serotonin reuptake inhibitors and mirtazapine have been recommended for depression, while atypical antipsychotics are recommended for psychosis and behavioral problems.[81] Specialist neuropsychiatric input is recommended since people may require long-term treatment with multiple medications in combination.[26]
Plant-based medications
[edit]There has been a number of alternative therapies experimented in ayurvedic medicine with plant-based products, although none have provided good evidence of efficacy. A recent study showed that the stromal processing peptidase (SPP), a synthetic enzyme found in plant chloroplasts, prevented the aggregation of proteins associated with Huntington's disease.[91] However, repeat studies and clinical validation are needed to confirm its true therapeutic potential.
Education
[edit]The families of individuals, and society at large, who have inherited or are at risk of inheriting HD have generations of experience of HD but may be unaware of recent breakthroughs in understanding the disease, and of the availability of genetic testing. Genetic counseling benefits these individuals by updating their knowledge, seeking to dispel any unfounded beliefs that they may have, and helping them consider their future options and plans. The Patient Education Program for Huntington's Disease has been created to help educate family members, caretakers, and those diagnosed with Huntington's disease.[92] Also covered is information concerning family planning choices, care management, and other considerations.[26][93]
Prognosis
[edit]The length of the trinucleotide repeat accounts for 60% of the variation of the age of symptoms onset and their rate of progress. A longer repeat results in an earlier age of onset and a faster progression of symptoms.[26][94] Individuals with more than sixty repeats often develop the disease before age 20, while those with fewer than 40 repeats may remain asymptomatic.[95] The remaining variation is due to environmental factors and other genes that influence the mechanism of the disease.[26]
Life expectancy in HD is generally around 10 to 30 years following the onset of visible symptoms.[26] Juvenile Huntington's disease has a life expectancy rate of 10 years after onset of visible symptoms. Most life-threatening complications result from muscle coordination, and to a lesser extent, behavioral changes induced by declining cognitive function. The largest risk is pneumonia, which causes death in one third of those with HD. As the ability to synchronize movements deteriorates, difficulty clearing the lungs, and an increased risk of aspirating food or drink both increase the risk of contracting pneumonia. The second-greatest risk is heart disease, which causes almost a quarter of fatalities of those with HD.[96] Suicide is the third greatest cause of fatalities, with 7.3% of those with HD taking their own lives and up to 27% attempting to do so. To what extent suicidal thoughts are influenced by behavioral symptoms is unclear, as they signify a desire to avoid the later stages of the disease.[97][98][99] Suicide is the greatest risk of this disease before the diagnosis is made and in the middle stages of development throughout the disease. Other associated risks include choking; due to the inability to swallow, physical injury from falls, and malnutrition.[96][20]
Epidemiology
[edit]The late onset of Huntington's disease means it does not usually affect reproduction.[26] The worldwide prevalence of HD is 5–10 cases per 100,000 persons,[100][101] but varies greatly geographically as a result of ethnicity, local migration and past immigration patterns.[26] Prevalence is similar for men and women. The rate of occurrence is highest in peoples of Western European descent, averaging around seven per 100,000 people, and is lower in the rest of the world; e.g., one per million people of Asian and African descent. A 2013 epidemiological study of the prevalence of Huntington's disease in the UK between 1990 and 2010 found that the average prevalence for the UK was 12.3 per 100,000.[26][102] Additionally, some localized areas have a much higher prevalence than their regional average.[26] One of the highest incidences is in the isolated populations of the Lake Maracaibo region of Venezuela, where HD affects up to 700 per 100,000 persons.[26][103] Other areas of high localization have been found in Tasmania and specific regions of Scotland, Wales and Sweden.[99] Increased prevalence in some cases occurs due to a local founder effect, a historical migration of carriers into an area of geographic isolation.[99][104] Some of these carriers have been traced back hundreds of years using genealogical studies.[99] Genetic haplotypes can also give clues for the geographic variations of prevalence.[99][105] Iceland, on the contrary, has a rather low prevalence of 1 per 100,000, despite the fact that Icelanders as a people are descended from the early Germanic tribes of Scandinavia which also gave rise to the Swedes; all cases with the exception of one going back nearly two centuries having derived from the offspring of a couple living early in the 19th century.[106] Finland, as well, has a low incidence of only 2.2 per 100,000 people.[107]
Until the discovery of a genetic test, statistics could only include clinical diagnosis based on physical symptoms and a family history of HD, excluding those who died of other causes before diagnosis. These cases can now be included in statistics; and, as the test becomes more widely available, estimates of the prevalence and incidence of the disorder are likely to increase.[99][108]
History
[edit]
In centuries past, various kinds of chorea were at times called by names such as Saint Vitus' dance, with little or no understanding of their cause or type in each case.
Первое определенное упоминание о ГБ было в письме Чарльза Оскара Уотерса (1816–1892), опубликованном в первом издании « Данглисона Робли Практики медицины» в 1842 году. [110] Waters described "a form of chorea, vulgarly called magrums", including accurate descriptions of the chorea, its progression, and the strong heredity of the disease.[111] In 1846 Charles Rollin Gorman (1817–1879) observed how higher prevalence seemed to occur in localized regions.[112][111] Independently of Gorman and Waters, both students of Dunglison at Jefferson Medical College in Philadelphia,[113] Йохан Кристиан Лунд (1830–1906) также произвел раннее описание в 1860 году. [ 111 ] Он особо отметил, что в Сетесдалене , уединенной горной долине в Норвегии , высокая распространенность деменции была связана с паттерном подергивания двигательных расстройств, который передавался в семьях. [ 114 ]
Первое подробное описание болезни было сделано Джорджем Хантингтоном в 1872 году. Изучая совокупную историю болезни нескольких поколений семей, демонстрирующих схожие симптомы, он понял, что их состояния должны быть связаны между собой; в своей первой статье он представил подробное и точное определение болезни. Хантингтон описал точную картину наследования аутосомно-доминантного заболевания за несколько лет до повторного открытия учеными менделевского наследования .
О своей наследственной природе. Когда у одного или обоих родителей наблюдаются проявления болезни... один или несколько потомков почти всегда страдают от этой болезни... Но если случайно эти дети проживут жизнь без нее, нить оборвется и внуки и правнуки первых шейкеров могут быть уверены, что они свободны от этой болезни. [ 109 ] [ 115 ]
Сэр Уильям Ослер интересовался этим расстройством и хореей в целом и был впечатлен статьей Хантингтона, в которой говорилось: «В истории медицины есть несколько случаев, когда болезнь была описана более точно, более наглядно или более кратко». [ 116 ] [ 111 ] [ 117 ] Постоянный интерес Ослера к ГБ в сочетании с его влиянием в области медицины помог быстро распространить осведомленность и знания об этом заболевании среди медицинского сообщества. [ 111 ] Большой интерес проявили учёные Европы, в том числе Луи Теофиль Жозеф Ландузи , Дезире-Маглуар Бурневиль , Камилло Гольджи и Жозеф Жюль Дежерин , и до конца века большая часть исследований БГ имела европейское происхождение. [ 111 ] К концу XIX века исследования и отчеты о ГБ были опубликованы во многих странах, и это заболевание было признано заболеванием во всем мире. [ 111 ]
Во время повторного открытия менделевского наследования на рубеже 20-го века БГ экспериментально использовалась как пример аутосомно-доминантного наследования. [ 111 ] Английский биолог Уильям Бейтсон использовал родословные затронутых семей, чтобы установить, что HD имеет аутосомно-доминантный тип наследования. [ 118 ] [ 113 ] Сильный характер наследования побудил нескольких исследователей, в том числе Смита Эли Джеллиффа , попытаться отследить и связать членов семьи, участвовавших в предыдущих исследованиях. [ 111 ] Джеллифф собрал информацию со всего Нью-Йорка и опубликовал несколько статей, касающихся генеалогии ГБ в Новой Англии . [ 119 ] Исследование Джеллиффа вызвало интерес у его друга по колледжу Чарльза Дэвенпорта , который поручил Элизабет Манси провести первое полевое исследование семей с HD на восточном побережье Соединенных Штатов и составить их родословные. [ 120 ] Давенпорт использовал эту информацию, чтобы документировать различный возраст начала и диапазон симптомов ГБ; он утверждал, что большинство случаев ГБ в США можно отнести к небольшому числу людей. [ 120 ] Это исследование было дополнительно приукрашено в 1932 году П.Р. Весси , который популяризировал идею о том, что три брата, покинувшие Англию в 1630 году и направлявшиеся в Бостон, были прародителями ГБ в США. [ 121 ] Утверждение о том, что были установлены самые ранние прародители, и евгеническая предвзятость работ Манси, Давенпорта и Весси способствовали недопониманию и предрассудкам в отношении БГ. [ 113 ] Манси и Давенпорт также популяризировали идею о том, что в прошлом некоторые люди с HD могли считаться одержимыми духами или жертвами колдовства , и иногда их избегали или изгоняли общество. [ 122 ] [ 123 ] Эта идея не доказана. Исследователи нашли противоположные доказательства; например, сообщество семьи, которую изучал Джордж Хантингтон, открыто принимало тех, у кого были симптомы ГБ. [ 113 ] [ 122 ]
Поиск причины этого состояния значительно активизировался в 1968 году, когда Фонд наследственных заболеваний был создан Милтоном Векслером , психоаналитиком из Лос-Анджелеса , Калифорния , у чьей жены Леоноры Сабин ранее в том же году была диагностирована болезнь Хантингтона, (HDF). . [ 124 ] Трое братьев жены Векслера также страдали этим заболеванием.
Фонд участвовал в привлечении более 100 ученых для участия в совместном проекте США и Венесуэлы по борьбе с болезнью Хантингтона, который в течение 10 лет, начиная с 1979 года, работал над выяснением генетической причины. [ 125 ] Это было достигнуто в 1983 году, когда был приблизительно обнаружен причинный ген, [ 104 ] а в 1993 году этот ген был точно расположен на хромосоме 4 (4p16.3). [ 126 ] Исследование было сосредоточено на населении двух изолированных венесуэльских деревень, Барранкитас и Лагунетас, где наблюдалась необычно высокая распространенность БГ, и охватило более 18 000 человек, в основном из одной большой семьи, и в результате БГ стал первым аутосомного заболевания. локусом обнаружено с помощью анализа генетического сцепления . [ 126 ] [ 127 ] Среди других инноваций в рамках проекта были разработаны методы маркировки ДНК , которые стали важным шагом на пути реализации проекта «Геном человека» . [ 125 ]
В то же время были сделаны ключевые открытия, касающиеся механизмов заболевания, в том числе выводы исследовательской группы Аниты Хардинг о влиянии длины гена. [ 128 ]
Моделирование заболевания на различных типах животных, таких как трансгенная мышь, выведенная в 1996 году, позволило провести более масштабные эксперименты. Поскольку у этих животных более быстрый метаболизм и гораздо более короткая продолжительность жизни, чем у людей, результаты экспериментов получаются раньше, что ускоряет исследования. Открытие в 1997 году того, что фрагменты mHtt неправильно сворачиваются, привело к открытию ядерных включений, которые они вызывают. Эти достижения привели к все более обширным исследованиям белков, связанных с этим заболеванием, потенциальных медикаментозных методов лечения, методов ухода и самого гена. [ 111 ] [ 129 ]
Сети ухода и поддержки, которые сложились в Венесуэле и Колумбии во время исследовательских проектов там в 1970-х и 2000-х годах, в конечном итоге были разрушены различными силами, такими как продолжающийся кризис в Венесуэле и смерть ведущего исследователя в Колумбии (Хорхе Даса Баррига). ). [ 130 ] Врачи работают над возрождением этих сетей, потому что люди, которые внесли вклад в науку о болезни Хантингтона, участвуя в этих исследованиях, заслуживают надлежащего последующего ухода; общества в других частях мира, которые извлекают выгоду из достигнутых таким образом научных достижений, обязаны по крайней мере этим тем, кто участвовал в исследованиях. [ 130 ]
Раньше это состояние называлось хореей Хантингтона, но этот термин был заменен болезнью Хантингтона, поскольку не у всех пациентов развивается хорея, а также из-за важности когнитивных и поведенческих проблем. [ 131 ]
Общество и культура
[ редактировать ]Этика
[ редактировать ]Генетическое тестирование болезни Хантингтона подняло несколько этических проблем. Вопросы генетического тестирования включают определение того, насколько зрелым должен быть человек, прежде чем его можно будет считать подходящим для тестирования, обеспечение конфиденциальности результатов и следует ли разрешить компаниям использовать результаты тестов для принятия решений о приеме на работу, страховании жизни или других финансовых вопросах. Возникли разногласия, когда Чарльз Дэвенпорт в 1910 году предложил использовать принудительную стерилизацию и иммиграционный контроль для людей с определенными заболеваниями, включая БГ, в рамках евгенического движения. [ 132 ] Экстракорпоральное оплодотворение имеет некоторые проблемы, связанные с использованием эмбрионов. Некоторые исследования БГ имеют этические проблемы из-за использования испытаний на животных и эмбриональных стволовых клеток . [ 133 ] [ 134 ]
Разработка точного диагностического теста на болезнь Хантингтона вызвала социальные, юридические и этические проблемы по поводу доступа к результатам человека и их использования. [ 135 ] [ 136 ] Многие руководства и процедуры тестирования предусматривают строгие процедуры раскрытия информации и конфиденциальности, позволяющие людям решать, когда и как получать результаты, а также кому они будут доступны. [ 26 ] Страховые компании и предприятия сталкиваются с вопросом, следует ли использовать результаты генетических тестов при оценке человека, например, при страховании жизни или трудоустройстве. Страховые компании Соединенного Королевства договорились с Министерством здравоохранения и социального обеспечения , что до 2017 года клиентам не нужно будет раскрывать им прогностические генетические тесты, но это соглашение прямо исключает одобренный правительством тест для болезни Хантингтона при оформлении полисов на сумму более 500 000 фунтов стерлингов. . [ 137 ] [ 138 ] Как и в случае с другими неизлечимыми генетическими заболеваниями с более поздним началом, проведение предсимптомного тестирования у ребенка или подростка является этически сомнительным, поскольку для этого человека не будет никакой медицинской пользы. Существует консенсус в отношении тестирования только лиц, которые считаются когнитивно зрелыми, хотя существует контраргумент, согласно которому родители имеют право принимать решение от имени своего ребенка. Из-за отсутствия эффективного лечения тестирование лица, не достигшего совершеннолетия , которое не признано дееспособным, в большинстве случаев считается неэтичным. [ 49 ] [ 139 ] [ 140 ]
Существуют этические проблемы, связанные с пренатальным генетическим тестированием или преимплантационной генетической диагностикой, чтобы гарантировать, что ребенок не родится с данным заболеванием. [ 141 ] Например, пренатальное тестирование поднимает вопрос о селективном аборте, который некоторые считают неприемлемым. [ 141 ] Поскольку это доминантное заболевание, возникают трудности в ситуациях, когда родитель не хочет знать свой диагноз. Это потребует сохранения частей процесса в секрете от родителя. [ 141 ]
Поддерживающие организации
[ редактировать ]
В 1968 году, после того как Милтон Векслер столкнулся с HD в семье своей жены, он был вдохновлен созданием Фонда наследственных заболеваний (HDF) с целью лечения генетических заболеваний путем координации и поддержки исследований. [ 17 ] Фонд и дочь Векслера, Нэнси Векслер , были ключевыми участниками исследовательской группы в Венесуэле, которая обнаружила ген HD. [ 17 ]
Примерно в то же время, когда сформировалась HDF, Марджори Гатри помогла основать комитет по борьбе с болезнью Хантингтона (ныне Американское общество по борьбе с болезнью Хантингтона ) после того, как ее муж, фолк-певец и автор песен Вуди Гатри умер от осложнений HD. [ 18 ]
С тех пор во многих странах мира сформировались организации поддержки и исследования, которые помогли повысить осведомленность общественности о БГ. Некоторые из них сотрудничают в зонтичных организациях, таких как Международная ассоциация Хантингтона и Европейская сеть HD. [ 142 ] Многие организации поддержки проводят ежегодные мероприятия по повышению осведомленности о БГ, некоторые из которых были одобрены соответствующими правительствами. Например, Сенат США объявил 6 июня «Национальным днем распространения информации о болезни Хантингтона» . [ 143 ] Существует множество организаций, которые поддерживают и информируют людей, страдающих ГБ, включая Ассоциацию болезни Хантингтона в Великобритании. Крупнейшим спонсором исследований является Фонд инициативы по лечению болезни Хантингтона (CHDI). [ 144 ]
Направления исследований
[ редактировать ]Исследования механизма ГБ сосредоточены на выявлении функционирования Htt, того, как mHtt отличается или мешает ему, а также патологии головного мозга, вызываемой этим заболеванием. [ 145 ] Исследования проводятся с использованием in vitro методов , генетически модифицированных животных (также называемых моделями трансгенных животных ) и людей-добровольцев. Модели на животных имеют решающее значение для понимания фундаментальных механизмов, вызывающих заболевание, а также для поддержки ранних стадий разработки лекарств . [ 129 ] Идентификация причинного гена позволила создать множество генетически модифицированных организмов , включая нематод (круглых червей), -дрозофил плодовых мух и генетически модифицированных млекопитающих, включая мышей, крыс, овец, свиней и обезьян, которые экспрессируют мутантный хантингтин и развивают прогрессирующую нейродегенерацию и HD-ген. как симптомы. [ 129 ]
В настоящее время проводятся исследования с использованием множества подходов, направленных либо на предотвращение болезни Хантингтона, либо на замедление ее прогрессирования. [ 145 ] Стратегии, модифицирующие заболевание, можно разделить на три категории: снижение уровня мутантного белка хантингтина (включая сплайсинг генов и подавление генов ); подходы, направленные на улучшение выживаемости нейронов за счет снижения вреда, наносимого белком конкретным клеточным путям и механизмам (включая гомеостаз белка и ингибирование деацетилазы гистонов ); и стратегии замены потерянных нейронов. Кроме того, разрабатываются новые методы лечения для улучшения функционирования мозга; они направлены на разработку симптоматических, а не модифицирующих заболевание методов лечения и включают ингибиторы фосфодиэстеразы . [ 146 ] [ 147 ]
Фонд CHDI финансирует множество исследовательских инициатив, публикуя множество публикаций. [ 148 ] Фонд CHDI является крупнейшим спонсором исследований болезни Хантингтона в мире и стремится найти и разработать лекарства, которые замедлят прогрессирование ГБ. [ 144 ] [ 149 ] CHDI ранее был известен как High Q Foundation. В 2006 году компания потратила 50 миллионов долларов на исследования болезни Хантингтона. [ 144 ] CHDI сотрудничает со многими академическими и коммерческими лабораториями по всему миру и занимается надзором и управлением исследовательскими проектами, а также финансированием. [ 150 ]
Сокращение производства хантингтина
[ редактировать ]Замалчивание генов направлено на снижение продукции мутантного белка, поскольку БХ вызывается единственным доминантным геном, кодирующим токсичный белок. Эксперименты по подавлению генов на моделях мышей показали, что когда экспрессия mHtt снижается, симптомы улучшаются. [ 151 ] Безопасность РНК-интерференции и методов подавления генов с помощью аллель-специфических олигонуклеотидов (ASO) была продемонстрирована на мышах и мозге более крупных приматов-макак. [ 152 ] [ 153 ] Аллель-специфическое подавление пытается заставить замолчать мутантный htt, оставляя Htt дикого типа нетронутым. Один из способов добиться этого — идентифицировать полиморфизмы, присутствующие только в одной аллели, и производить препараты, подавляющие гены, которые нацелены на полиморфизмы только в мутантном аллеле. [ 154 ] Первое испытание по подавлению генов с участием людей с БГ началось в 2015 году с целью проверки безопасности IONIS-HTTRx, произведенного Ionis Pharmaceuticals и проведенного Институтом неврологии UCL . [ 155 ] [ 156 ] Мутантный хантингтин был впервые обнаружен и количественно определен в спинномозговой жидкости носителей мутации болезни Хантингтона в 2015 году с использованием нового иммуноанализа «подсчет одиночных молекул» . [ 157 ] предоставляя прямой способ оценить, достигают ли методы снижения хантингтина желаемого эффекта. [ 158 ] [ 159 ] Испытание третьей фазы этого соединения, переименованного в томинерсен и спонсируемого Roche Pharmaceuticals , началось в 2019 году, но было остановлено в 2021 году после того, как совет по мониторингу безопасности пришел к выводу, что баланс риска и пользы был неблагоприятным. [ 160 ] Испытания генной терапии, снижающие хантингтин, проводимые Uniqure, начались в 2019 году, и было объявлено о нескольких испытаниях пероральных соединений-модуляторов сплайсинга, снижающих хантингтин. [ 161 ] сплайсинга генов Рассматриваются методы , чтобы попытаться восстановить геном с ошибочным геном, вызывающим БГ, с использованием таких инструментов, как CRISPR/Cas9 . [ 147 ]
Увеличение клиренса гентингтина
[ редактировать ]Другая стратегия снижения уровня мутантного хантингтина заключается в увеличении скорости, с которой клетки способны его выводить. [ 162 ] Поскольку mHtt (и многие другие белковые агрегаты ) разрушаются в результате аутофагии , увеличение скорости аутофагии потенциально может снизить уровни mHtt и тем самым облегчить течение заболевания. [ 163 ] Фармакологические и генетические индукторы аутофагии были протестированы на различных моделях болезни Хантингтона; Было показано, что многие из них снижают уровни mHtt и снижают токсичность. [ 162 ]
Улучшение выживаемости клеток
[ редактировать ]Среди подходов, направленных на улучшение выживаемости клеток в присутствии мутантного хантингтина, — коррекция регуляции транскрипции с помощью ингибиторов гистондеацетилазы , модуляция агрегации хантингтина, улучшение метаболизма и функции митохондрий и восстановление функции синапсов . [ 151 ]
Замена нейронов
[ редактировать ]Терапия стволовыми клетками используется для замены поврежденных нейронов путем трансплантации стволовых клеток в пораженные участки мозга. Эксперименты на животных моделях (только крысы и мыши) дали положительные результаты. [ 164 ]
Каким бы ни был их будущий терапевтический потенциал, стволовые клетки уже являются ценным инструментом для изучения болезни Хантингтона в лаборатории. [ 165 ]
Ферроптоз
[ редактировать ]Ферроптоз — это форма регулируемой гибели клеток, характеризующаяся зависимым от железа накоплением гидроперекисей липидов до летальных уровней. ALOX5 -опосредованный ферроптоз действует как путь гибели клеток при окислительном стрессе при болезни Хантингтона. [ 166 ] Ингибиторы ферроптоза защищают модели дегенеративных заболеваний головного мозга, в том числе Болезни Паркинсона, Хантингтона и Альцгеймера. [ 166 ]
Клинические испытания
[ редактировать ]В 2020 году было проведено 197 клинических исследований, связанных с различными методами лечения и биомаркерами болезни Хантингтона, которые были указаны как находящиеся в стадии реализации, набор или недавно завершенные. [ 167 ] соединения Испытанные , которые не смогли предотвратить или замедлить прогрессирование болезни Хантингтона, включают ремасемид , коэнзим Q10 , рилузол , креатин , миноциклин , этил-ЭПК , фенилбутират и димебон . [ 168 ]
См. также
[ редактировать ]Ссылки
[ редактировать ]- ^ Jump up to: а б с д и ж г час я дж к л Даялу П., Альбин Р.Л. (февраль 2015 г.). «Болезнь Гентингтона: патогенез и лечение». Неврологические клиники . 33 (1): 101–114. дои : 10.1016/j.ncl.2014.09.003 . ПМИД 25432725 .
- ^ Jump up to: а б с д и ж г Кэрон Н.С., Райт Дж.Э., Хайден М.Р. (2020). Адам М.П., Ардингер Х.Х., Пагон Р.А., Уоллес С.Е., Бин Л.Дж., Стивенс К., Амемия А. (ред.). «Болезнь Хантингтона». Джин Обзоры . ПМИД 20301482 .
- ^ Jump up to: а б с д и ж г час я дж к л м Фрэнк С. (январь 2014 г.). «Лечение болезни Гентингтона» . Нейротерапия . 11 (1): 153–160. дои : 10.1007/s13311-013-0244-z . ПМЦ 3899480 . ПМИД 24366610 .
- ^ Jump up to: а б с д и ж г час «Информационная страница о болезни Хантингтона» . Национальный институт неврологических расстройств и инсульта . Архивировано из оригинала 13 декабря 2020 года . Проверено 14 декабря 2020 г.
- ^ Jump up to: а б Дурр А., Гарджуло М., Фейнгольд Дж. (ноябрь 2012 г.). «Пресимптоматическая фаза болезни Хантингтона». Ревю Неврологии . 168 (11): 806–808. дои : 10.1016/j.neurol.2012.07.003 . ПМИД 22902173 .
- ^ Ферри ФФ (2010). Дифференциальный диагноз Ферри: практическое руководство по дифференциальной диагностике симптомов, признаков и клинических нарушений (2-е изд.). Филадельфия, Пенсильвания: Эльзевир/Мосби. п. Глава H. ISBN 978-0-323-07699-9 .
- ^ «Болезнь Хантингтона – Лечение и поддержка» . Национальная служба здравоохранения Великобритании . 23 октября 2017 года. Архивировано из оригинала 6 мая 2023 года . Проверено 6 мая 2023 г.
- ^ Jump up to: а б Illarioshkin SN, Klyushnikov SA, Vigont VA, Seliverstov YA, Kaznacheyeva EV (September 2018). "Molecular Pathogenesis in Huntington's Disease" . Biochemistry. Biokhimiia . 83 (9): 1030–1039. doi : 10.1134/S0006297918090043 . PMID 30472941 . S2CID 26471825 . Archived from the original on 13 November 2020 . Retrieved 8 November 2020 – via protein.bio.msu.ru.
- ^ Jump up to: а б с Судхакар В., Ричардсон Р.М. (январь 2019 г.). «Генная терапия нейродегенеративных заболеваний» . Нейротерапия . 16 (1): 166–175. дои : 10.1007/s13311-018-00694-0 . ПМК 6361055 . ПМИД 30542906 .
- ^ Jump up to: а б с Кумар, Аббас А., Астер Дж. (2018). Основная патология Роббинса (Десятое изд.). Филадельфия, Пенсильвания: Эльзевир. п. 879. ИСБН 978-0-323-35317-5 .
- ^ Первс Д. (2012). Нейронаука (5-е изд.). Сандерленд, Массачусетс: Sinauer Associates. п. 415. ИСБН 978-0-87893-695-3 .
- ^ Jump up to: а б с Сауду Ф., Умберт С. (март 2016 г.). «Биология Хантингтина» . Нейрон . 89 (5): 910–926. дои : 10.1016/j.neuron.2016.02.003 . ПМИД 26938440 . S2CID 8272667 .
- ^ Jump up to: а б с д и Бейтс Г.П., Дорси Р., Гуселла Дж.Ф., Хайден М.Р., Кей С., Ливитт Б.Р. и др. (апрель 2015 г.). «Болезнь Хантингтона». Обзоры природы. Праймеры по болезням . 1 : 15005. дои : 10.1038/nrdp.2015.5 . ПМИД 27188817 . S2CID 25759303 .
- ^ «Аспирационная пневмония: что это такое, причины, диагностика, лечение» . Кливлендская клиника . Архивировано из оригинала 12 июня 2023 года . Проверено 12 июня 2023 г.
- ^ Jump up to: а б Вейл ТК, Кардосо Ф (2015). «Хорея: Путешествие в историю» . Тремор и другие гиперкинетические движения . 5 . дои : 10.7916/D8WM1C98 . ПМЦ 4454991 . ПМИД 26056609 .
- ^ Jump up to: а б «О болезни Хантингтона» . Genome.gov . Архивировано из оригинала 9 января 2021 года . Проверено 13 января 2021 г.
- ^ Jump up to: а б с д «История ХДФ» . Фонд наследственных болезней. Архивировано из оригинала 19 ноября 2015 года . Проверено 18 ноября 2015 г.
- ^ Jump up to: а б «История и генетика болезни Хантингтона | Американское общество болезни Хантингтона» . Март 2019. Архивировано из оригинала 1 декабря 2020 года . Проверено 14 декабря 2020 г.
- ^ Jump up to: а б Йенсен Р.Н., Болвиг Т., Соренсен С.А. (март 2018 г.). «[Психические симптомы у пациентов с болезнью Хантингтона]». Ugeskrift для Laeger (на датском языке). 180 (13). ПМИД 29587954 .
- ^ Jump up to: а б «Болезнь Хантингтона – Симптомы и причины» . Клиника Мэйо . Архивировано из оригинала 5 марта 2018 года . Проверено 13 декабря 2022 г.
- ^ «Болезнь Хантингтона» . www.nhsinform.scot. Архивировано из оригинала 12 июля 2020 года . Проверено 12 июля 2020 г.
- ^ Jump up to: а б с Кэрон Н.С., Райт Дж.Э., Хайден М.Р. (июнь 2020 г.). «Болезнь Хантингтона» . Книжная полка Genereviews . Университет Вашингтона. ПМИД 20301482 . Архивировано из оригинала 10 февраля 2009 года . Проверено 22 ноября 2020 г.
- ^ Диагностическое и статистическое руководство по психическим расстройствам: DSM-5 (5-е изд.). Арлингтон, Вирджиния: Американская психиатрическая ассоциация. 2013. с. 639. ИСБН 978-0-89042-554-1 .
- ^ Кремер Б. (2002). «Клиническая неврология болезни Хантингтона». В Бейтс Г., Харпер П., Джонс Л. (ред.). Болезнь Хантингтона – Третье издание . Оксфорд: Издательство Оксфордского университета. стр. 28–53. ISBN 978-0-19-851060-4 .
- ^ Wagle AC, Wagle SA, Маркова И.С., Берриос Г.Е. (2000). «Психиатрическая заболеваемость при болезни Гентингтона». Неврология, психиатрия и исследования мозга (8): 5–16.
- ^ Jump up to: а б с д и ж г час я дж к л м н тот п д р с т в v В х и С аа аб и объявление но из в ах есть также и аль являюсь а к ап ак с как в В из хорошо топор является тот нет бб до нашей эры др. быть парень бг чб Уокер Ф.О. (январь 2007 г.). «Болезнь Хантингтона». Ланцет . 369 (9557): 218–228. дои : 10.1016/S0140-6736(07)60111-1 . ПМИД 17240289 . S2CID 46151626 .
- ^ Кремер Б. (2002). «Клиническая неврология болезни Хантингтона». В Бейтс Г., Харпер П., Джонс Л. (ред.). Болезнь Хантингтона – Третье издание . Оксфорд: Издательство Оксфордского университета. стр. 28–53. ISBN 978-0-19-851060-4 .
- ^ Jump up to: а б с д Монтойя А., Прайс Б.Х., Менеар М., Лепаж М. (январь 2006 г.). «Визуализация мозга и когнитивные дисфункции при болезни Хантингтона» (PDF) . Журнал психиатрии и неврологии . 31 (1): 21–29. ПМК 1325063 . ПМИД 16496032 . Архивировано из оригинала (PDF) 23 марта 2016 года . Проверено 17 сентября 2008 г.
- ^ Jump up to: а б Дики А.С., Ла Спада, Арканзас (апрель 2018 г.). «Развитие терапии болезни Гентингтона: от текущих стратегий к новым возможностям» . Американский журнал медицинской генетики. Часть А. 176 (4): 842–861. дои : 10.1002/ajmg.a.38494 . ПМЦ 5975251 . ПМИД 29218782 .
- ^ Азиз Н.А., ван дер Марк М.А., Пейл Х., Олде Риккерт М.Г., Блум Б.Р., Роос Р.А. (декабрь 2008 г.). «Потеря веса при нейродегенеративных заболеваниях». Журнал неврологии . 255 (12): 1872–1880. дои : 10.1007/s00415-009-0062-8 . ПМИД 19165531 . S2CID 26109381 .
- ^ «Брошюра Канадского общества Хантингтона» (PDF) . Справочник для лиц, осуществляющих уход за болезнью Хантингтона на поздней стадии . Общество HD Канады. 11 апреля 2007 г. Архивировано из оригинала (PDF) 25 июня 2008 г. . Проверено 10 августа 2008 г.
- ^ Jump up to: а б с ван Дуйн Э., Кингма Э.М., ван дер Маст Р.К. (2007). «Психопатология у подтвержденных носителей гена болезни Хантингтона». Журнал нейропсихиатрии и клинических нейронаук . 19 (4): 441–448. дои : 10.1176/appi.neuropsych.19.4.441 . ПМИД 18070848 .
- ^ Мюррей Э.Д., Баттнер Н., Прайс Б.Х. (2012). «Депрессия и психоз в неврологической практике». В Брэдли В.Г., Дарофф Р.Б., Феничел Г.М., Янкович Дж. (ред.). Неврология Брэдли в клинической практике (6-е изд.). Филадельфия, Пенсильвания: Эльзевир/Сондерс. п. 108. ИСБН 978-1-4377-0434-1 .
- ^ ван дер Бург Дж. М., Бьоркквист М., Брундин П. (август 2009 г.). «За пределами мозга: широко распространенная патология при болезни Хантингтона». «Ланцет». Неврология . 8 (8): 765–774. дои : 10.1016/S1474-4422(09)70178-4 . ПМИД 19608102 . S2CID 14419437 .
- ^ Кацуно М., Банно Х., Сузуки К., Такеучи Ю., Кавасима М., Танака Ф. и др. (май 2008 г.). «Молекулярная генетика и биомаркеры полиглутаминовых заболеваний». Современная молекулярная медицина . 8 (3): 221–234. дои : 10.2174/156652408784221298 . ПМИД 18473821 .
- ^ Сквитьери Ф, Фрати Л, Чармиелло А, Ластория С, Куоррел О (февраль 2006 г.). «Ювенильная болезнь Хантингтона: отличается ли дозо-эффект патогенный механизм от классической болезни взрослых?». Механизмы старения и развития . 127 (2): 208–212. дои : 10.1016/j.mad.2005.09.012 . ПМИД 16274727 . S2CID 20523093 .
- ^ Нэнс М.А., Майерс Р.Х. (2001). «Ювенильное начало болезни Хантингтона - клинические и исследовательские перспективы». Обзоры исследований умственной отсталости и нарушений развития . 7 (3): 153–157. дои : 10.1002/mrdd.1022 . ПМИД 11553930 .
- ^ Пассаж Е (2001). Цветной атлас генетики (2-е изд.). Темы. п. 142 . ISBN 978-0-86577-958-7 .
- ^ «Секс связан» . Genome.gov . Архивировано из оригинала 14 апреля 2022 года . Проверено 13 декабря 2022 г.
- ^ Ридли Р.М., Фрит К.Д., Кроу Т.Дж., Коннели П.М. (сентябрь 1988 г.). «Предчувствие при болезни Гентингтона наследуется по мужской линии, но может возникать и по женской линии» . Журнал медицинской генетики . 25 (9): 589–595. дои : 10.1136/jmg.25.9.589 . ПМЦ 1051535 . ПМИД 2972838 .
- ^ Семака А., Крейтон С., Уорби С., Хайден М.Р. (октябрь 2006 г.). «Прогностическое тестирование на болезнь Гентингтона: интерпретация и значение промежуточных аллелей». Клиническая генетика . 70 (4): 283–294. дои : 10.1111/j.1399-0004.2006.00668.x . ПМИД 16965319 . S2CID 26007984 .
- ^ Векслер Н.С., Янг А.Б., Танци Р.Э., Трэверс Х., Староста-Рубинштейн С., Пенни Дж.Б. и др. (1987). «Гомозиготы по болезни Гентингтона». Природа . 326 (6109): 194–197. Бибкод : 1987Natur.326..194W . дои : 10.1038/326194a0 . hdl : 2027.42/62543 . ПМИД 2881213 . S2CID 4312171 .
- ^ Сквитьери Ф., Геллера С., Каннелла М., Мариотти С., Цислаги Г., Рубинштейн Д.С. и др. (апрель 2003 г.). «Гомозиготность по мутации CAG при болезни Хантингтона связана с более тяжелым клиническим течением» . Мозг . 126 (Часть 4): 946–955. дои : 10.1093/brain/awg077 . ПМИД 12615650 .
- ^ Гёлер Х., Лаловски М., Стельцль У., Вельтер С., Стродике М., Ворм У. и др. (сентябрь 2004 г.). «Сеть взаимодействия белков связывает GIT1, усилитель агрегации хантингтина, с болезнью Хантингтона» . Молекулярная клетка . 15 (6): 853–865. doi : 10.1016/j.molcel.2004.09.016 . ПМИД 15383276 .
- ^ Глайч К.Е., Садри-Вакили Г (2015). «Эпигенетические механизмы, участвующие в патогенезе болезни Хантингтона» . Журнал болезни Хантингтона . 4 (1): 1–15. дои : 10.3233/JHD-159001 . ПМИД 25813218 .
- ^ Jump up to: а б Лю З, Чжоу Т, Зиглер А.С., Димитрион П., Цзо Л. (2017). «Окислительный стресс при нейродегенеративных заболеваниях: от молекулярных механизмов к клиническому применению» . Окислительная медицина и клеточное долголетие . 2017 : 2525967. дои : 10.1155/2017/2525967 . ПМК 5529664 . ПМИД 28785371 .
- ^ Jump up to: а б с Каттанео Э., Зуккато С., Тартари М. (декабрь 2005 г.). «Нормальная функция хантингтина: альтернативный подход к болезни Гентингтона». Обзоры природы. Нейронаука . 6 (12): 919–930. дои : 10.1038/nrn1806 . ПМИД 16288298 . S2CID 10119487 .
- ^ Jump up to: а б с д и Рубинштейн, округ Колумбия, Кармайкл Дж (август 2003 г.). «Болезнь Хантингтона: молекулярная основа нейродегенерации». Обзоры экспертов в области молекулярной медицины . 5 (20): 1–21. дои : 10.1017/S1462399403006549 . ПМИД 14585171 . S2CID 28435830 .
- ^ Jump up to: а б Блох М., Хайден М.Р. (январь 1990 г.). «Мнение: прогностическое тестирование на болезнь Хантингтона в детстве: проблемы и последствия» . Американский журнал генетики человека . 46 (1): 1–4. ПМК 1683548 . ПМИД 2136787 .
- ^ Матлахов И., ван дер Вель ПК (декабрь 2019 г.). «Конформационные исследования патогенных расширенных полиглутаминовых белковых отложений при болезни Хантингтона» . Экспериментальная биология и медицина . 244 (17): 1584–1595. дои : 10.1177/1535370219856620 . ПМК 6920524 . ПМИД 31203656 . S2CID 189944779 .
- ^ Jump up to: а б с Садри-Вакили Г., Ча Дж. Х. (июнь 2006 г.). «Механизмы заболевания: модификации гистонов при болезни Хантингтона». Природная клиническая практика. Неврология . 2 (6): 330–338. дои : 10.1038/ncpneuro0199 . ПМИД 16932577 . S2CID 12474262 .
- ^ Кумар А., Ратан Р.Р. (октябрь 2016 г.). «Окислительный стресс и болезнь Хантингтона: хорошее, плохое и злое» . Журнал болезни Хантингтона . 5 (3): 217–237. дои : 10.3233/JHD-160205 . ПМК 5310831 . ПМИД 27662334 .
- ^ ПК Нопулос (март 2016 г.). «Болезнь Хантингтона: моногенное дегенеративное заболевание полосатого тела» . Диалоги в клинической неврологии . 18 (1): 91–98. doi : 10.31887/DCNS.2016.18.1/pnopoulos . ПМЦ 4826775 . ПМИД 27069383 .
- ^ МакКолган П., Тебризи С.Дж. (январь 2018 г.). «Болезнь Хантингтона: клинический обзор» . Европейский журнал неврологии . 25 (1): 24–34. дои : 10.1111/ene.13413 . ПМИД 28817209 .
- ^ Первс Д., Августин Г.А., Фитцпатрик Д., Холл В., ЛаМантия А.С., Макнамара Д.О. и др. (2001). «Модуляция движения базальных ганглиев - цепи в системе базальных ганглиев» . В Первес Д. (ред.). Нейронаука (2-е изд.). Сандерленд, Массачусетс: Sinauer Associates. ISBN 978-0-87893-742-4 . Архивировано из оригинала 18 февраля 2009 года . Проверено 1 апреля 2009 г.
- ^ Лобсигер CS, Кливленд DW (ноябрь 2007 г.). «Глиальные клетки как внутренние компоненты неклеточных автономных нейродегенеративных заболеваний» . Природная неврология . 10 (11): 1355–1360. дои : 10.1038/nn1988 . ПМК 3110080 . ПМИД 17965655 .
- ^ Jump up to: а б Кроссман А.Р. (май 2000 г.). «Функциональная анатомия двигательных нарушений» . Журнал анатомии . 196 (Часть 4): 519–525. дои : 10.1046/j.1469-7580.2000.19640519.x . ПМК 1468094 . ПМИД 10923984 .
- ^ Даффи Дж (2013). Двигательные нарушения речи: субстраты, дифференциальный диагноз и лечение (3-е изд.). Сент-Луис, Миссури: Эльзевир. стр. 196–7.
- ^ Стеффан Дж.С., Бодай Л., Паллос Дж., Поулман М., МакКэмпбелл А., Апостол Б.Л. и др. (октябрь 2001 г.). «Ингибиторы гистондеацетилазы останавливают полиглутамин-зависимую нейродегенерацию у дрозофилы» . Природа . 413 (6857): 739–743. Бибкод : 2001Natur.413..739S . дои : 10.1038/35099568 . ПМИД 11607033 . S2CID 4419980 . Архивировано из оригинала 1 августа 2020 года . Проверено 28 июня 2019 г.
- ^ Петрушка Дж., Хартенстайн М.Дж., Гудман М.Ф. (февраль 1998 г.). «Анализ проскальзывания цепи в расширениях ДНК-полимеразы триплетных повторов CAG/CTG, связанных с нейродегенеративными заболеваниями» . Журнал биологической химии . 273 (9): 5204–5210. дои : 10.1074/jbc.273.9.5204 . ПМИД 9478975 .
- ^ Гайяр Ф (1 мая 2007 г.). «Болезнь Хантингтона» . Радиологическая картина дня . www.radpod.org. Архивировано из оригинала 22 октября 2007 года . Проверено 24 июля 2009 г.
- ^ Рао АК, Муратори Л, Луи ЭД, Московиц КБ, Мардер КС (апрель 2009 г.). «Клиническое измерение нарушений подвижности и равновесия при болезни Гентингтона: достоверность и оперативность». Походка и осанка . 29 (3): 433–436. дои : 10.1016/j.gaitpost.2008.11.002 . ПМИД 19111470 .
- ^ «Единая шкала оценки болезни Хантингтона (UHDRS)» . UHDRS и база данных . ГСГ. 1 февраля 2009 года. Архивировано из оригинала 11 августа 2015 года . Проверено 14 апреля 2009 г.
- ^ Майерс Р.Х. (апрель 2004 г.). «Генетика болезни Хантингтона» . НейроРкс . 1 (2): 255–262. дои : 10.1602/neurorx.1.2.255 . ПМК 534940 . ПМИД 15717026 .
- ^ Jump up to: а б с д и ж г де Ди-Смолдерс CE, де Верт GM, Либерс И, Тиббен А, Эверс-Кибумс Г (май 2013 г.). «Репродуктивные возможности будущих родителей в семьях с болезнью Гентингтона: клинические, психологические и этические размышления» . Обновление репродукции человека . 19 (3): 304–315. дои : 10.1093/humupd/dms058 . ПМИД 23377865 . де Ди-Смолдерс CE, де Верт GM, Либерс И, Тиббен А, Эверс-Кибумс Г (2013). «Репродуктивные возможности будущих родителей в семьях с болезнью Гентингтона: клинические, психологические и этические размышления» . Обновление репродукции человека . 19 (3): 304–315. дои : 10.1093/humupd/dms058 . ПМИД 23377865 .
- ^ Байг С.С., Стронг М., Россер Э., Тавернер Н.В., Глю Р., Медзибродска З. и др. (октябрь 2016 г.). «22 года прогностического тестирования болезни Хантингтона: опыт Британского консорциума по прогнозированию Хантингтона» . Европейский журнал генетики человека . 24 (10): 1396–1402. дои : 10.1038/ejhg.2016.36 . ПМК 5027682 . ПМИД 27165004 .
- ^ Форрест Кинан К., Симпсон С.А., Медзибродска З., Александр Д.А., Семпер Дж. (июнь 2013 г.). «Как партнеры узнают о риске болезни Хантингтона в парных отношениях?». Журнал генетического консультирования . 22 (3): 336–344. дои : 10.1007/s10897-012-9562-2 . ПМИД 23297124 . S2CID 15447709 .
- ^ Эрвин С., Уильямс Дж.К., Джул А.Р., Менгелинг М., Миллс Дж.А., Бомбард Ю. и др. (июль 2010 г.). «Восприятие, опыт и реакция на генетическую дискриминацию при болезни Хантингтона: международное исследование RESPOND-HD» . Американский журнал медицинской генетики. Часть B. Нейропсихиатрическая генетика . 153Б (5): 1081–1093. дои : 10.1002/ajmg.b.31079 . ПМЦ 3593716 . ПМИД 20468061 .
- ^ Берсон CM, Марки КР (сентябрь 2001 г.). «Проблемы генетического консультирования при прогностическом генетическом тестировании семейных неврологических заболеваний, возникающих во взрослом возрасте». Семинары по детской неврологии . 8 (3): 177–186. дои : 10.1053/spen.2001.26451 . ПМИД 11575847 .
- ^ Смит Дж. А., Мичи С., Стивенсон М., Куоррел О. (март 2002 г.). «Восприятие риска и процессы принятия решений у кандидатов на генетическое тестирование на болезнь Гентингтона: интерпретирующий феноменологический анализ». Журнал психологии здоровья . 7 (2): 131–144. дои : 10.1177/1359105302007002398 . ПМИД 22114233 . S2CID 40182214 .
- ^ Jump up to: а б Хайден М.Р. (март 2003 г.). «Прогностическое тестирование на болезнь Хантингтона: универсальная модель?». «Ланцет». Неврология . 2 (3): 141–142. дои : 10.1016/S1474-4422(03)00317-X . ПМИД 12849232 . S2CID 39581496 .
- ^ «Руководство по прогностическому тесту молекулярной генетики при болезни Хантингтона. Международная ассоциация Хантингтона (IHA) и Всемирная федерация неврологов (WFN) Исследовательская группа по хорее Хантингтона». Неврология . 44 (8): 1533–1536. Август 1994 г. doi : 10.1212/WNL.44.8.1533 . ПМИД 8058167 . S2CID 28018134 .
- ^ Лосекут М., ван Бельзен М.Дж., Сенека С., Бауэр П., Стенхаус С.А., Бартон Д.Э. (май 2013 г.). «Руководство по передовому опыту EMQN/CMGS по молекулярно-генетическому тестированию болезни Хантингтона» . Европейский журнал генетики человека . 21 (5): 480–486. дои : 10.1038/ejhg.2012.200 . ПМЦ 3641377 . ПМИД 22990145 .
- ^ Шульман Дж.Д., Блэк С.Х., Хэндисайд А., Нэнс В.Е. (февраль 1996 г.). «Преимплантационное генетическое тестирование на болезнь Хантингтона и некоторые другие доминантно наследуемые заболевания». Клиническая генетика . 49 (2): 57–58. дои : 10.1111/j.1399-0004.1996.tb04327.x . ПМИД 8740912 . S2CID 45703511 .
- ^ Стерн Х.Дж., Хартон Г.Л., Сиссон М.Е., Джонс С.Л., Фэллон Л.А., Торселл Л.П. и др. (июнь 2002 г.). «Неразглашаемая преимплантационная генетическая диагностика болезни Хантингтона». Пренатальная диагностика . 22 (6): 503–507. дои : 10.1002/pd.359 . ПМИД 12116316 . S2CID 33967835 .
- ^ «Прогностическое тестирование на болезнь Хантингтона» . 2011. Архивировано из оригинала 22 января 2013 года . Проверено 7 мая 2013 г.
- ^ Кулиев А, Верлинский Ю (апрель 2005 г.). «Преимплантационная диагностика: реалистичный вариант вспомогательной репродукции и генетической практики». Современное мнение в области акушерства и гинекологии . 17 (2): 179–183. дои : 10.1097/01.gco.0000162189.76349.c5 . ПМИД 15758612 . S2CID 9382420 .
- ^ «Руководство по генетическому тестированию болезни Хантингтона» . Фонд наследственных болезней. Архивировано из оригинала 26 июня 2015 года . Проверено 7 мая 2013 г.
- ^ Jump up to: а б Шнайдер С.А., Уокер Р.Х., Бхатия К.П. (сентябрь 2007 г.). «Синдромы, подобные болезни Хантингтона: что следует учитывать у пациентов с отрицательным тестом на ген болезни Хантингтона». Природная клиническая практика. Неврология . 3 (9): 517–525. дои : 10.1038/ncpneuro0606 . ПМИД 17805246 . S2CID 9052603 .
- ^ Фрэнк С., Янкович Дж. (март 2010 г.). «Достижения в фармакологическом лечении болезни Хантингтона» . Наркотики . 70 (5): 561–571. дои : 10.2165/11534430-000000000-00000 . ПМИД 20329804 . S2CID 42386743 . Архивировано из оригинала 8 октября 2011 года.
- ^ Jump up to: а б с Бонелли Р.М., Веннинг Г.К., Капфхаммер Х.П. (март 2004 г.). «Болезнь Хантингтона: современные методы лечения и будущие методы лечения». Международная клиническая психофармакология . 19 (2): 51–62. дои : 10.1097/00004850-200403000-00001 . ПМИД 15076012 . S2CID 1956458 .
- ^ Панагиотакис П.Х., ДиСарио Дж.А., Хильден К., Огара М., Фанг Дж.К. (2008). «Размещение трубки DPEJ предотвращает аспирационную пневмонию у пациентов из группы высокого риска». Питание в клинической практике . 23 (2): 172–175. дои : 10.1177/0884533608314537 . ПМИД 18390785 .
- ^ Jump up to: а б «Руководство по физиотерапии EHDN» (PDF) . Европейская рабочая группа по сетевой физиотерапии HD. Архивировано из оригинала (PDF) 4 марта 2016 года . Проверено 15 ноября 2015 г.
- ^ Куин Л., Буси М. (февраль 2012 г.). «Разработка рекомендаций по физиотерапии и классификаций лечения людей с болезнью Гентингтона» . Управление нейродегенеративными заболеваниями . 2 (1): 21–31. дои : 10.2217/nmt.11.86 . Архивировано из оригинала 9 августа 2020 года . Проверено 10 мая 2012 г.
- ^ Халил Х., Куинн Л., ван Дёрсен Р., Мартин Р., Россер А., Бусс М. (январь 2012 г.). «Приверженность использованию DVD с домашними упражнениями у людей с болезнью Гентингтона: точки зрения участников» . Физиотерапия . 92 (1): 69–82. дои : 10.2522/ptj.20100438 . ПМИД 21960468 .
- ^ Трэверс Э., Джонс К., Никол Дж. (март 2007 г.). «Оказание паллиативной помощи при болезни Хантингтона». Международный журнал паллиативного ухода . 13 (3): 125–130. дои : 10.12968/ijpn.2007.13.3.23274 . ПМИД 17505405 .
- ^ «FDA одобрило первый препарат для лечения хореи при болезни Гентингтона» . Управление по контролю за продуктами и лекарствами США. 15 августа 2008 года. Архивировано из оригинала 21 августа 2008 года . Проверено 10 августа 2008 г.
- ^ Коппен Э.М., Роос Р.А. (январь 2017 г.). «Современные фармакологические подходы к уменьшению хореи при болезни Хантингтона» . Наркотики . 77 (1): 29–46. дои : 10.1007/s40265-016-0670-4 . ПМК 5216093 . ПМИД 27988871 .
- ^ Морси С., Халил С.М., Дохейм М.Ф., Камель М.Г., Эль-Басиони Д.С., Ахмед Хасан Х.И. и др. (август 2019 г.). «Эффективность этил-ЭПК в лечении болезни Хантингтона: систематический обзор и метаанализ». Acta Neuropsychiatrica . 31 (4): 175–185. дои : 10.1017/neu.2019.11 . hdl : 10069/39427 . ПМИД 30890195 . S2CID 84183892 .
- ^ Центр исследований по оценке лекарств (17 июля 2019 г.). «В поисках поздней дискинезии: революционное определение и одобрение валбеназина» . FDA . Архивировано из оригинала 3 декабря 2020 года . Проверено 15 ноября 2020 г.
- ^ Лламас Э., Коюнку С., Ли Х.Дж., Верманн М., Гутьеррес-Гарсия Р., Данкен Н. и др. (ноябрь 2023 г.). «Экспрессия in planta человеческого полиQ-расширенного фрагмента хантингтина обнаруживает механизмы предотвращения агрегации белков, связанных с заболеванием» . Природное старение . 3 (11): 1345–1357. дои : 10.1038/s43587-023-00502-1 . ПМЦ 10645592 . ПМИД 37783816 .
- ^ А'Кампо Л.Е., Сплиетхофф-Камминга Н.Г., Роос Р.А. (2012). «Программа обучения пациентов по поводу болезни Гентингтона (ПКП-ГД)» . Дж. Хантингтонс Дис . 1 (1): 47–56. дои : 10.3233/JHD-2012-120002 . ПМИД 25063190 .
- ^ Харпер П. (2002). «Генетическое консультирование и предсимптомное тестирование». В Бейтс Г., Харпер П., Джонс Л. (ред.). Болезнь Хантингтона – Третье издание . Оксфорд: Издательство Оксфордского университета. стр. 198–242. ISBN 978-0-19-851060-4 .
- ^ Харпер П.С. (июнь 1999 г.). «Болезнь Хантингтона: клиническая, генетическая и молекулярная модель нарушений полиглутаминовых повторов» . Философские труды Лондонского королевского общества. Серия Б, Биологические науки . 354 (1386): 957–61. дои : 10.1098/rstb.1999.0446 . ПМЦ 1692597 . ПМИД 10434293 .
- ^ Эндрю С.Е., Голдберг Ю.П., Кремер Б., Телениус Х., Тейлманн Дж., Адам С. и др. (август 1993 г.). «Взаимосвязь между длиной повторов тринуклеотида (CAG) и клиническими особенностями болезни Хантингтона». Природная генетика . 4 (4): 398–403. дои : 10.1038/ng0893-398 . ПМИД 8401589 . S2CID 20645822 .
- ^ Jump up to: а б Уокер Ф.О. (январь 2007 г.). «Болезнь Хантингтона». Ланцет . 369 (9557): 218–28. дои : 10.1016/S0140-6736(07)60111-1 . ПМИД 17240289 . S2CID 46151626 .
- ^ Кроуфорд Д., Сноуден Дж. (2002). «Нейропсихологические и нервно-психические аспекты болезни Хантингтона». В Бейтс Г., Харпер П., Джонс Л. (ред.). Болезнь Хантингтона – Третье издание . Оксфорд: Издательство Оксфордского университета. стр. 62–87. ISBN 978-0-19-851060-4 .
- ^ Ди Майо Л., Сквитьери Ф., Наполитано Дж., Кампанелла Дж., Трофаттер Дж.А., Коннелли П.М. (апрель 1993 г.). «Суицидальный риск при болезни Хантингтона» . Журнал медицинской генетики . 30 (4): 293–5. дои : 10.1136/jmg.30.4.293 . ПМЦ 1016335 . ПМИД 8487273 .
- ^ Jump up to: а б с д и ж Харпер П. (2002). «Эпидемиология болезни Хантингтона». В Бейтс Г., Харпер П., Джонс Л. (ред.). Болезнь Хантингтона – Третье издание . Оксфорд: Издательство Оксфордского университета. стр. 159–189. ISBN 978-0-19-851060-4 .
- ^ Шарон И., Шэрон Р., Уилкенс Дж.П., Эрсан Т. (2010). «Деменция болезни Хантингтона» . медицина, WebMD . Медскейп. Архивировано из оригинала 5 марта 2010 года . Проверено 16 мая 2010 г.
- ^ Драйвер-Данкли Э., Кэвинесс Дж. Н. (2007). «Болезнь Хантингтона». В Шапира А.Х. (ред.). Неврология и клиническая неврология . Мосби Эльзевир. стр. 879–885. ISBN 978-0-323-03354-1 .
- ^ Эванс С.Дж., Дуглас И., Роулинз М.Д., Векслер Н.С., Тебризи С.Дж., Смит Л. (октябрь 2013 г.). «Распространенность болезни Хантингтона у взрослых в Великобритании на основе диагнозов, зафиксированных в записях общей практики» . Журнал неврологии, нейрохирургии и психиатрии . 84 (10): 1156–60. дои : 10.1136/jnnp-2012-304636 . ПМЦ 3786631 . ПМИД 23482661 .
- ^ Авила-Жироо Р. (1973). «Медицинские и социальные аспекты хореи Хантингтона в штате Сулия, Венесуэла». Достижения в неврологии . 1 : 261–6. ISSN 0091-3952 . НАИД 10021247802.
- ^ Jump up to: а б Гуселла Дж.Ф., Векслер Н.С., Коннели П.М., Нейлор С.Л., Андерсон М.А., Танзи Р.Э. и др. (1983). «Полиморфный ДНК-маркер, генетически связанный с болезнью Хантингтона». Природа . 306 (5940): 234–8. Бибкод : 1983Natur.306..234G . дои : 10.1038/306234a0 . ПМИД 6316146 . S2CID 4320711 .
- ^ Сквитьери Ф., Эндрю С.Е., Голдберг Ю.П., Кремер Б., Спенс Н., Зейслер Дж. и др. (декабрь 1994 г.). «Анализ гаплотипов ДНК болезни Хантингтона открывает ключ к разгадке истоков и механизмов распространения CAG, а также причин географических различий распространенности». Молекулярная генетика человека . 3 (12): 2103–14. дои : 10.1093/hmg/3.12.2103 . ПМИД 7881406 .
- ^ Свейнссон О, Халлдорссон С, Олафссон Э (июль 2012 г.). «Необычайно низкая распространенность болезни Хантингтона в Исландии». Европейская неврология . 68 (1): 48–51. дои : 10.1159/000337680 . ПМИД 22722209 . S2CID 207551998 .
- ^ Сипиля Ю., Хиетала М., Сийтонен А., Пяйваринта М., Маямаа К. (январь 2015 г.). «Эпидемиология болезни Гентингтона в Финляндии». Паркинсонизм и связанные с ним расстройства . 21 (1): 46–9. дои : 10.1016/j.parkreldis.2014.10.025 . ПМИД 25466405 .
- ^ Альмквист Э.В., Элтерман Д.С., Маклеод П.М., Хайден М.Р. (сентябрь 2001 г.). «Высокий уровень заболеваемости и отсутствие семейного анамнеза у четверти пациентов, у которых впервые диагностирована болезнь Хантингтона в Британской Колумбии». Клиническая генетика . 60 (3): 198–205. дои : 10.1034/j.1399-0004.2001.600305.x . ПМИД 11595021 . S2CID 19786394 .
- ^ Jump up to: а б Хантингтон Дж. (1872 г.). «О Хорея» . Медицинский и хирургический репортер Филадельфии . 26 (15): 317–321. ISBN 978-90-6186-011-2 . Архивировано из оригинала 21 апреля 2022 года . Проверено 21 апреля 2022 г.
- ^ Данглисон Р. (1842 г.). Медицинская практика... Том. 2. Филадельфия, Пенсильвания, США: Леа и Бланшар. стр. 312–313. Архивировано из оригинала 20 апреля 2022 года . Проверено 20 апреля 2022 г.
- ^ Jump up to: а б с д и ж г час я дж Харпер П. (2002). «Болезнь Хантингтона: историческая справка». В Бейтс Г., Харпер П., Джонс Л. (ред.). Болезнь Хантингтона – Третье издание . Оксфорд: Издательство Оксфордского университета. стр. 3–24. ISBN 978-0-19-851060-4 .
- ^ Горман CR (1846 г.). О форме хореи, в просторечии называемой Magrums (докторская диссертация). Филадельфия: Медицинский колледж Джефферсона.
- «Каталог Медицинского колледжа Джефферсона в Филадельфии: сессия 1846–1847 годов» . п. 14. Архивировано из оригинала 26 июня 2022 года.
- Данглисон Р. (1848). Медицинская практика... Том. 2 (3-е изд.). Филадельфия, Пенсильвания, США: Леа и Бланшар. п. 218.
- Бойд В.А. (6 ноября 1913 г.). «Наследственная хорея с сообщением о случае» . Бостонский медицинский и хирургический журнал . 169 (19): 680–685. дои : 10.1056/NEJM191311061691904 . Архивировано из оригинала 2 августа 2023 года . Проверено 21 апреля 2022 г. См. стр. 680.
- ^ Jump up to: а б с д Векслер А, Векслер Н (2008). Женщина, вошедшая в море. Хантингтон и возникновение генетической болезни . Издательство Йельского университета. п. 288. ИСБН 978-0-300-10502-5 . Проверено 15 ноября 2015 г.
- ^ Лунд Дж. К. (1860). «Хорея Сти Вити в Сетерсдалене. Отрывок из медицинского отчета окружного доктора Дж. К. Лунда за 1860 год». Отчет о состоянии здоровья : 137–138.
- ^ Ланска диджей (апрель 2000 г.). «Джордж Хантингтон (1850–1916) и наследственная хорея». Журнал истории нейронаук . 9 (1): 76–89. дои : 10.1076/0964-704X(200004)9:1;1-2;FT076 . ПМИД 11232352 . S2CID 22659368 .
- ^ Ослер В. (1908). «Исторические заметки о наследственной хорее» . Нейрографы . 1 (2): 113–116. Архивировано из оригинала 21 апреля 2022 года . Проверено 21 апреля 2022 г. См. стр. 115.
- См. также: Ослер В. (февраль 1893 г.). «Замечания о разновидностях хронической хореи и отчет о двух семьях наследственной формы с одним аутопсией» . Журнал нервных и психических заболеваний . 20 (2): 97–111. Архивировано из оригинала 21 апреля 2022 года . Проверено 21 апреля 2022 г. См. стр. 97.
- ^ Броуди И.А., Уилкинс Р.Х. (сентябрь 1967 г.). «Хорея Хантингтона». Архив неврологии . 17 (3): 331. doi : 10.1001/archneur.1967.00470270109013 . ПМИД 4228262 .
- ^ Бейтсон В. (1909). Принципы наследственности Менделя . Кембридж, Англия: Издательство Кембриджского университета. п. 229. Бейтсон называет «болезнь Хантингтона» «наследственной хореей».
- ^ Джеллифф С.Э., Манси Э.Б., Давенпорт CB (1913). «Хорея Хантингтона: исследование наследственности» . Журнал нервных и психических заболеваний . 40 (12): 796–799. дои : 10.1097/00005053-191312000-00010 . Архивировано из оригинала 5 апреля 2022 года . Проверено 19 сентября 2020 г.
- ^ Jump up to: а б Давенпорт CB, Манси EB (1916). «Хорея Хантингтона в связи с наследственностью и евгеникой» . Американский журнал безумия . 73 (2): 195–222. дои : 10.1176/ajp.73.2.195 .
- ^ Весси П.Р. (1932). «О передаче хореи Гентингтона за 300 лет – группа семьи Бурес» . Нервные и психические заболевания . 76 (6): 553–573. дои : 10.1097/00005053-193212000-00001 . S2CID 147656032 . Архивировано из оригинала 28 августа 2021 года . Проверено 1 апреля 2009 г.
- ^ Jump up to: а б Векслер А.Р. (2002). «Хорея и община в городе девятнадцатого века». Бюллетень истории медицины . 76 (3): 495–527. дои : 10.1353/bhm.2002.0150 . ПМИД 12486915 . S2CID 30791504 .
- ^ Коннели ПМ (май 1984 г.). «Болезнь Гентингтона: генетика и эпидемиология» . Американский журнал генетики человека . 36 (3): 506–26. ПМК 1684448 . ПМИД 6233902 .
- ^ Векслер Н.С. (2012). «Болезнь Хантингтона: пропаганда, движущая наука» . Ежегодный обзор медицины . 63 : 1–22. doi : 10.1146/annurev-med-050710-134457 . ПМИД 22248319 .
- ^ Jump up to: а б «Венесуэльский проект по болезни Хантингтона» . Сайт Фонда наследственных заболеваний . Фонд наследственных болезней. 2008. Архивировано из оригинала 10 августа 2015 года . Проверено 8 сентября 2008 г.
- ^ Jump up to: а б «Новый ген, содержащий тринуклеотидный повтор, который расширен и нестабильен на хромосомах при болезни Хантингтона. Совместная исследовательская группа по болезни Хантингтона». Клетка . 72 (6): 971–983. Март 1993 г. doi : 10.1016/0092-8674(93)90585-E . hdl : 2027.42/30901 . ПМИД 8458085 . S2CID 802885 .
- ^ Бертрам Л., Танзи Р.Э. (июнь 2005 г.). «Генетическая эпидемиология нейродегенеративных заболеваний» . Журнал клинических исследований . 115 (6): 1449–1457. дои : 10.1172/JCI24761 . ПМК 1137006 . ПМИД 15931380 .
- ^ Ла Спада А.Р., Ролинг Д.Б., Хардинг А.Е., Уорнер К.Л., Шпигель Р., Хаусманова-Петрусевич I и др. (декабрь 1992 г.). «Мейотическая стабильность и генотип-фенотипическая корреляция тринуклеотидного повтора при Х-сцепленной спинальной и бульбарной мышечной атрофии». Природная генетика . 2 (4): 301–4. дои : 10.1038/ng1292-301 . ПМИД 1303283 . S2CID 6603129 .
- ^ Jump up to: а б с Росс К.А., Тебризи С.Дж. (январь 2011 г.). «Болезнь Хантингтона: от молекулярного патогенеза к клиническому лечению». «Ланцет». Неврология . 10 (1): 83–98. дои : 10.1016/S1474-4422(10)70245-3 . ПМИД 21163446 . S2CID 17488174 .
- ^ Jump up to: а б Смит Дж. Э., Кордеро К. (23 мая 2023 г.). «Разыскал наука, а потом забыл» . Нью-Йорк Таймс . Архивировано из оригинала 23 мая 2023 года . Проверено 23 мая 2023 г.
- ^ «Что такое HD?» . Ассоциация болезни Гентингтона. Архивировано из оригинала 13 декабря 2011 года . Проверено 18 декабря 2011 г.
- ^ Давенпорт CB (май 1915 г.). «Хорея Хантингтона в связи с наследственностью и евгеникой» . Труды Национальной академии наук Соединенных Штатов Америки . 1 (5): 283–5. Бибкод : 1915PNAS....1..283D . дои : 10.1073/pnas.1.5.283 . ПМЦ 1090803 . ПМИД 16575999 .
- ^ Роллин Б.Е. (2006). «Регулирование исследований на животных и возникновение этики животных: концептуальная история» (PDF) . Теоретическая медицина и биоэтика . 27 (4): 285–304. дои : 10.1007/s11017-006-9007-8 . ПМИД 16937023 . S2CID 18620094 . Архивировано из оригинала (PDF) 8 октября 2020 года . Проверено 1 декабря 2019 г.
- ^ Дорфлингер Р.М. (февраль 2008 г.). «Проблема обмана в исследованиях эмбриональных стволовых клеток» . Пролиферация клеток . 41 (Приложение 1): 65–70. дои : 10.1111/j.1365-2184.2008.00492.x . ПМК 6496399 . ПМИД 18181947 .
- ^ Чепмен М.А. (июль 1990 г.). «Прогностическое тестирование генетических заболеваний, возникающих у взрослых: этические и юридические последствия использования анализа сцепления для болезни Гентингтона» . Американский журнал генетики человека . 47 (1): 1–3. ПМЦ 1683745 . ПМИД 2140926 .
- ^ Хаггинс М., Блох М., Канани С., Куоррел О.В., Тейлман Дж., Хедрик А. и др. (июль 1990 г.). «Этические и юридические дилеммы, возникающие во время прогностического тестирования заболеваний, начинающихся у взрослых: опыт болезни Хантингтона» . Американский журнал генетики человека . 47 (1): 4–12. ПМЦ 1683755 . ПМИД 1971997 .
- ^ «Мораторий на страхование генетики продлен до 2017 года» (Пресс-релиз). Ассоциация британских страховщиков. 5 апреля 2011 года. Архивировано из оригинала 4 марта 2016 года . Проверено 13 января 2016 г.
- ^ «Эксперт поддерживает раскрытие результатов генных тестов» . Статья Би-би-си. 7 июня 2007 г. Архивировано из оригинала 26 февраля 2008 г.
- ^ Бинеделл Дж., Солдан-младший, Скурфилд Дж., Харпер П.С. (ноябрь 1996 г.). «Прогностическое тестирование болезни Хантингтона: аргументы в пользу подхода к оценке запросов подростков» . Журнал медицинской генетики . 33 (11): 912–8. дои : 10.1136/jmg.33.11.912 . ПМЦ 1050784 . ПМИД 8950670 .
- ^ Борри П., Гоффин Т., Найс Х., Дирикс К. (2008). «Прогностическое генетическое тестирование несовершеннолетних на генетические заболевания, возникающие во взрослом возрасте» . Медицинский журнал Mount Sinai, Нью-Йорк . 75 (3): 287–96. дои : 10.1002/msj.20038 . ПМИД 18704981 .
- ^ Jump up to: а б с Брауде П.Р., Де Верт ГМ, Эверс-Кибумс Дж., Петтигрю Р.А., Гераедтс Дж.П. (декабрь 1998 г.). «Преимплантационная генетическая диагностика болезни Хантингтона с неразглашением: практические и этические дилеммы». Пренатальная диагностика . 18 (13): 1422–6. doi : 10.1002/(SICI)1097-0223(199812)18:13<1422::AID-PD499>3.0.CO;2-R . ПМИД 9949442 . S2CID 39977672 .
- ^ «Международная ассоциация Хантингтона» . Международная ассоциация Хантингтона. 2013. Архивировано из оригинала 18 апреля 2009 года . Проверено 3 апреля 2009 г.
- ^ «Резолюция Сената США 531» . С. Рез. 531 . Сенат США. 6 апреля 2008 г. Архивировано из оригинала 17 ноября 2015 г. . Проверено 10 августа 2008 г.
- ^ Jump up to: а б с Одлинг-Сми Л. (17 мая 2007 г.). «Биомедицинская филантропия: денежное дерево» . Природа . 447 (7142): 251. Бибкод : 2007Natur.447..251. . дои : 10.1038/447251a . S2CID 4357517 .
- ^ Jump up to: а б «Болезнь Хантингтона: надежда через исследования» . www.ninds.nih.gov . Архивировано из оригинала 25 октября 2020 года . Проверено 16 ноября 2020 г.
- ^ Wild EJ, Тебризи SJ (сентябрь 2014 г.). «Цели будущих клинических исследований болезни Хантингтона: что находится в стадии разработки?» . Двигательные расстройства . 29 (11): 1434–45. дои : 10.1002/mds.26007 . ПМК 4265300 . ПМИД 25155142 .
- ^ Jump up to: а б Соломон С. (3 мая 2017 г.). «Таубе выделит 3 миллиона долларов на исследования болезни Хантингтона в США» . Таймс Израиля . Архивировано из оригинала 3 мая 2017 года . Проверено 5 мая 2017 г.
- ^ «Научные публикации | Фонд CHDI» . chdifoundation.org . Архивировано из оригинала 12 декабря 2021 года . Проверено 12 декабря 2021 г.
- ^ «Фонд ЧДИ» . chdifoundation.org . Архивировано из оригинала 14 ноября 2020 года . Проверено 13 ноября 2020 г.
- ^ Проверьте E (май 2007 г.). «Биомедицинская филантропия: любовь или деньги» . Природа . 447 (7142): 252–3. Бибкод : 2007Natur.447..252C . дои : 10.1038/447252a . ПМИД 17507955 . S2CID 4318384 .
- ^ Jump up to: а б Муньос-Санджуан I, врач Бейтса (февраль 2011 г.). «Важность интеграции фундаментальных и клинических исследований для разработки новых методов лечения болезни Хантингтона» . Журнал клинических исследований . 121 (2): 476–83. дои : 10.1172/JCI45364 . ПМК 3026740 . ПМИД 21285520 .
- ^ МакБрайд Дж.Л., Питцер М.Р., Будро Р.Л., Дюфур Б., Хоббс Т., Охеда С.Р. и др. (декабрь 2011 г.). «Доклиническая безопасность РНКи-опосредованного подавления HTT у макак-резус как потенциального лечения болезни Хантингтона» . Молекулярная терапия . 19 (12): 2152–62. дои : 10.1038/mt.2011.219 . ПМЦ 3242667 . ПМИД 22031240 .
- ^ Кордасевич Х.Б., Станек Л.М., Ванцевич Е.В., Мазур С., Макалонис М.М., Пытел К.А. и др. (июнь 2012 г.). «Устойчивое терапевтическое обращение болезни Хантингтона за счет временного подавления синтеза хантингтина» . Нейрон . 74 (6): 1031–44. дои : 10.1016/j.neuron.2012.05.009 . ПМЦ 3383626 . ПМИД 22726834 .
- ^ Барнс Д.В., Уитли Р.Дж. (февраль 1987 г.). «Противовирусная терапия и легочные заболевания». Грудь . 91 (2): 246–51. дои : 10.1378/сундук.91.2.246 . ПМИД 3026739 .
- ^ «Начинается суд над Лэндмарком Хантингтоном» . Архивировано из оригинала 21 октября 2015 года . Проверено 19 октября 2015 г.
- ^ «Безопасность, переносимость, фармакокинетика и фармакодинамика IONIS-HTTRx у пациентов с ранней манифестацией болезни Хантингтона - полный текст» . ClinicalTrials.gov. Архивировано из оригинала 29 сентября 2015 года . Проверено 18 апреля 2016 г.
- ^ Уайлд Э.Дж., Богджио Р., Лангбен Д., Робертсон Н., Хайдер С., Миллер Дж.Р. и др. (май 2015 г.). «Количественное определение мутантного белка хантингтина в спинномозговой жидкости пациентов с болезнью Гентингтона» . Журнал клинических исследований . 125 (5): 1979–86. дои : 10.1172/jci80743 . ПМЦ 4463213 . ПМИД 25844897 .
- ^ Чейз А (май 2015 г.). «Болезнь Хантингтона: спинномозговая жидкость и биомаркеры МРТ для продромальной HD». Обзоры природы Неврология . 11 (5): 245. doi : 10.1038/nrneurol.2015.63 . ПМИД 25896083 . S2CID 38300571 .
- ^ Кейзер М.С., Кордасевич Х.Б., Макбрайд Дж.Л. (апрель 2016 г.). «Стратегии подавления генов при доминантно наследственных нейродегенеративных заболеваниях: уроки болезни Хантингтона и спиноцеребеллярной атаксии» . Молекулярная генетика человека . 25 (Р1): Р53–64. дои : 10.1093/hmg/ddv442 . ПМЦ 4802374 . ПМИД 26503961 .
- ^ Квон Д. (6 апреля 2021 г.). «Генетическая терапия дает новую надежду на борьбу с неизлечимыми заболеваниями головного мозга». Природа . 592 (7853): 180–183. Бибкод : 2021Natur.592..180K . дои : 10.1038/d41586-021-00870-x . ПМИД 33824521 . S2CID 233173862 .
- ^ Хардинг Р. (26 апреля 2021 г.). Фокс Л. (ред.). «Обзор клинических исследований болезни Хантингтона» . HDBuzz . Архивировано из оригинала 5 мая 2021 года . Проверено 5 мая 2021 г.
- ^ Jump up to: а б Джаджадикерта А., Кешри С., Павел М., Престил Р., Райан Л., Рубинштейн, округ Колумбия (апрель 2020 г.). «Индукция аутофагии как терапевтическая стратегия нейродегенеративных заболеваний» . Журнал молекулярной биологии . Аутофагия при нейродегенеративных заболеваниях. 432 (8): 2799–2821. дои : 10.1016/j.jmb.2019.12.035 . ПМИД 31887286 . S2CID 209518157 . Архивировано из оригинала 23 мая 2022 года . Проверено 20 июня 2023 г.
- ^ Равикумар Б., Вашер С., Бергер З., Дэвис Дж.Э., Луо С., Ороз Л.Г. и др. (июнь 2004 г.). «Ингибирование mTOR вызывает аутофагию и снижает токсичность экспансии полиглутамина на моделях болезни Хантингтона на мухах и мышах» . Природная генетика . 36 (6): 585–595. дои : 10.1038/ng1362 . ПМИД 15146184 . S2CID 7749825 .
- ^ Холли С.М., Камджу Т., Рейдлинг Дж.К., Фьюри Б., Коул-Бергум Д., Бауэр Г. и др. (апрель 2018 г.). «Терапевтические эффекты стволовых клеток на моделях болезни Хантингтона на грызунах: обзор и электрофизиологические данные» . Нейронауки и терапия ЦНС . 24 (4). Уайли: 329–342. дои : 10.1111/cns.12839 . ПМК 6489814 . ПМИД 29512295 .
- ^ Кандифф П.Е., Андерсон С.А. (июнь 2011 г.). «Влияние индуцированных плюрипотентных стволовых клеток на изучение заболеваний центральной нервной системы» . Текущее мнение в области генетики и развития . 21 (3): 354–61. дои : 10.1016/j.где.2011.01.008 . ПМЦ 3932563 . ПМИД 21277194 .
- ^ Jump up to: а б Стоквелл Б.Р., Фридман Анджели Дж.П., Байир Х., Буш А.И., Конрад М., Диксон С.Дж. и др. (октябрь 2017 г.). «Ферроптоз: регулируемая связь клеточной смерти, связывающая метаболизм, окислительно-восстановительную биологию и болезни» . Клетка . 171 (2): 273–285. дои : 10.1016/j.cell.2017.09.021 . ПМЦ 5685180 . ПМИД 28985560 .
- ^ «Поиск: Болезнь Хантингтона — Результаты списка — ClinicalTrials.gov» . www.clinicaltrials.gov . Архивировано из оригинала 11 ноября 2021 года . Проверено 16 ноября 2020 г.
- ^ «Завершенные клинические испытания» . Исследовательская группа Хантингтона. Архивировано из оригинала 28 июня 2012 года . Проверено 4 февраля 2012 г.
Внешние ссылки
[ редактировать ]- Болезнь Хантингтона у Керли
- Проект HOPES. Архивировано 27 августа 2020 года в Wayback Machine - информационный проект Стэнфордского университета в формате HD.
- HDBuzz – новости исследований HD, написанные учеными простым языком
- HD Drug Works – новости о текущих методах лечения и запланированных исследованиях
- болезнь Хантингтона
- Аутосомно-доминантные заболевания
- Расстройства, вызывающие судороги
- Болезни, названные в честь первооткрывателей
- Экстрапирамидные и двигательные расстройства
- Генетические заболевания и расстройства
- Системные атрофии, поражающие преимущественно центральную нервную систему.
- Нарушения тринуклеотидных повторов