ЕСЛИ
| Боковой амиотрофический склероз | |
|---|---|
| Другие имена |
|
 | |
| Parts of the nervous system affected by ALS, causing progressive symptoms in skeletal muscles throughout the body[2] | |
| Specialty | Neurology |
| Symptoms | Early: Stiff muscles, muscle twitches, gradual increasing weakness[3] Later: Difficulty in speaking, swallowing, and breathing; respiratory failure;[3] 10–15% experience frontotemporal dementia[2] |
| Complications | Falling (accident); Respiratory failure; Pneumonia; Malnutrition |
| Usual onset | 45–75 years[2] |
| Causes | Unknown (about 85%), genetic (about 15%) |
| Risk factors | Genetic risk factors; age; male sex; heavy metals; organic chemicals; smoking; electric shock; physical exercise; head injury[2] |
| Diagnostic method | Clinical diagnosis of exclusion based on progressive symptoms of upper and lower motor neuron degeneration in which no other explanation can be found. Supportive evidence from electromyography, genetic testing, and neuroimaging |
| Differential diagnosis | Multifocal motor neuropathy, Kennedy's disease, Hereditary spastic paraplegia, Nerve compression syndrome, Diabetic neuropathy, Post-polio syndrome, Myasthenia gravis, Multiple sclerosis[4] |
| Treatment | Walker (mobility); Wheelchair; Non-invasive ventilation;[5] Feeding tube; Augmentative and alternative communication; symptomatic management |
| Medication | Riluzole, Edaravone, Sodium phenylbutyrate/ursodoxicoltaurine, Tofersen, Dextromethorphan/quinidine |
| Prognosis | Life expectancy highly variable but typically 2–4 years after diagnosis[6] |
| Frequency | |
Боковой амиотрофический склероз ( БАС как болезнь двигательных нейронов ( БДН ) или болезнь Лу Герига ), также известный в США , является редким терминальным нейродегенеративным заболеванием , которое приводит к прогрессирующей потере как верхних, так и нижних двигательных нейронов , которые обычно контролируют произвольные мышцы. сокращение. [3] БАС является наиболее распространенной формой заболеваний двигательных нейронов . [8] БАС часто проявляется на ранних стадиях постепенной мышечной ригидностью , подергиваниями , слабостью и истощением . [3] Потеря двигательных нейронов обычно продолжается до тех пор, пока не будут утрачены способности есть, говорить, двигаться и, наконец, дышать. [3] Хотя только у 15% людей с БАС также полностью развивается лобно-височная деменция , по оценкам, 50% сталкиваются, по крайней мере, с некоторыми незначительными трудностями с мышлением и поведением . [9] В зависимости от того, какой из вышеупомянутых симптомов развивается первым, БАС классифицируется как начало в конечностях (начинается со слабости в руках или ногах) или бульбарное начало (начинается с затруднений при разговоре или глотании ). [10]
Most cases of ALS (about 90–95%) have no known cause, and are known as sporadic ALS.[3][11] However, both genetic and environmental factors are believed to be involved.[12] The remaining 5–10% of cases have a genetic cause, often linked to a history of the disease in the family, and these are known as familial ALS (hereditary).[6][13] About half of these genetic cases are due to disease-causing variants in one of four specific genes.[14] The diagnosis is based on a person's signs and symptoms, with testing conducted to rule out other potential causes.[3]
There is no known cure for ALS.[3] The goal of treatment is to slow the disease progression, and improve symptoms.[9] Treatments that slow ALS include riluzole (extends life by two to three months) and sodium phenylbutyrate/ursodoxicoltaurine (extends life by around seven months).[15][16] Non-invasive ventilation may result in both improved quality, and length of life.[5] Mechanical ventilation can prolong survival but does not stop disease progression.[17] A feeding tube may help maintain weight and nutrition.[18] Death is usually caused by respiratory failure.[19] The disease can affect people of any age, but usually starts around the age of 60.[19] The average survival from onset to death is two to four years, though this can vary, and about 10% of those affected survive longer than ten years.[20]
Descriptions of the disease date back to at least 1824 by Charles Bell.[21] In 1869, the connection between the symptoms and the underlying neurological problems was first described by French neurologist Jean-Martin Charcot, who in 1874 began using the term amyotrophic lateral sclerosis.[21]
Classification
[edit]ALS is a motor neuron disease, which is a group of neurological disorders that selectively affect motor neurons, the cells that control voluntary muscles of the body.[3] Other motor neuron diseases include primary lateral sclerosis (PLS), progressive muscular atrophy (PMA), progressive bulbar palsy, pseudobulbar palsy, and monomelic amyotrophy (MMA).[22]
As a disease, ALS itself can be classified in a few different ways: by which part of the motor neurons are affected; by the parts of the body first affected; whether it is genetic; and the age at which it started. Each individual diagnosed with the condition will sit at a unique place at the intersection of these complex and overlapping subtypes, which presents a challenge to diagnosis, understanding, and prognosis.[23]
Subtypes of motor neuron disease
[edit]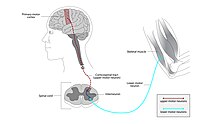
ALS can be classified by the types of motor neurons that are affected.[2] To successfully control any voluntary muscle in the body, a signal must be sent from the motor cortex in the brain down the upper motor neuron as it travels down the spinal cord. There, it connects via a synapse to the lower motor neuron which connects to the muscle itself. Damage to either the upper or lower motor neuron, as it makes its way from the brain to muscle, causes different types of symptoms.[24] Damage to the upper motor neuron typically causes spasticity including stiffness and increased tendon reflexes, and/or clonus, while damage to the lower motor neuron typically causes weakness, muscle atrophy, and fasciculations.[25]
Classical, or classic ALS, involves degeneration to both the upper motor neurons in the brain and the lower motor neurons in the spinal cord.[6][2] Primary lateral sclerosis (PLS) involves degeneration of only the upper motor neurons, and progressive muscular atrophy (PMA) involves only the lower motor neurons. There is debate over whether PLS and PMA are separate diseases or simply variants of ALS.[9]
| Main ALS Subtypes | Upper motor neuron degeneration | Lower motor neuron degeneration |
|---|---|---|
| Classical ALS | Yes | Yes |
| Primary lateral sclerosis (PLS) | Yes | No |
| Progressive muscular atrophy (PMA) | No | Yes |
Classical ALS accounts for about 70% of all cases of ALS and can be subdivided into where symptoms first appear as these are usually focussed to one region of the body at initial presentation before later spread. Limb-onset ALS (also known as spinal-onset) and bulbar-onset ALS.[9] Limb-onset ALS begins with weakness in the hands, arms, feet, and/or legs[10] and accounts for about two-thirds of all classical ALS cases.[9] Bulbar-onset ALS begins with weakness in the muscles of speech, chewing, and swallowing[24] and accounts for about 25% of classical ALS cases.[6] A rarer type of classical ALS affecting around 3% of patients is respiratory-onset,[9] in which the initial symptoms are difficulty breathing (dyspnea) upon exertion, at rest, or while lying flat (orthopnea).[26]
Primary lateral sclerosis (PLS) is a subtype of the overall ALS category which accounts for about 5% of all cases and only affects the upper motor neurons in the arms, legs, and bulbar region.[27] However, more than 75% of people with apparent PLS go on to later develop lower motor neuron signs within four years of symptom onset, meaning that a definitive diagnosis of PLS cannot be made until several years have passed.[28] PLS has a better prognosis than classical ALS, as it progresses slower, results in less functional decline, does not affect the ability to breathe, and causes less severe weight loss than classical ALS.[27]
Progressive muscular atrophy (PMA) is another subtype that accounts for about 5% of the overall ALS category and affects lower motor neurons in the arms, legs, and bulbar region.[27] While PMA is associated with longer survival on average than classical ALS, it is still progressive over time, eventually leading to respiratory failure and death.[9] As with PLS developing into classical ALS, PMA can also develop into classical ALS over time if the lower motor neuron involvement progresses to include upper motor neurons, in which case the diagnosis might be changed to classic ALS.[28]
Rare isolated variants of ALS
[edit]Isolated variants of ALS have symptoms that are limited to a single region for at least a year; they progress more slowly than classical ALS and are associated with longer survival.[2] These regional variants of ALS can only be considered as a diagnosis should the initial symptoms fail to spread to other spinal cord regions for an extended period of time (at least 12 months).[29] Flail arm syndrome is characterized by lower motor neuron damage affecting the arm muscles, typically starting with the upper arms symmetrically and progressing downwards to the hands.[2] Flail leg syndrome is characterized by lower motor neuron damage leading to asymmetrical weakness and wasting in the legs starting around the feet.[2] Isolated bulbar palsy is characterized by upper or lower motor neuron damage in the bulbar region (in the absence of limb symptoms for at least 20 months),[30] leading to gradual onset of difficulty with speech (dysarthria) and swallowing (dysphagia).

Age of onset
[edit]ALS can also be classified based on the age of onset. While the peak age of onset is 58 to 63 for sporadic ALS and 47 to 52 for genetic ALS,[19] about 10% of all cases of ALS begin before age 45 ("young-onset" ALS), and about 1% of all cases begin before age 25 ("juvenile" ALS).[24] People who develop young-onset ALS are more likely to be male, less likely to have bulbar onset of symptoms, and more likely to have a slower progression of the disease.[28] Juvenile ALS is more likely to be genetic in origin than adult-onset ALS; the most common genes associated with juvenile ALS are FUS, ALS2, and SETX.[31] Although most people with juvenile ALS live longer than those with adult-onset ALS, some of them have specific mutations in FUS and SOD1 that are associated with a poor prognosis.[32] Late onset (after age 65) is generally associated with a more rapid functional decline and shorter survival.[33]
Signs and symptoms
[edit]The disorder causes muscle weakness, atrophy, and muscle spasms throughout the body due to the degeneration of the upper motor and lower motor neurons. Sensory nerves and the autonomic nervous system are generally unaffected, meaning the majority of people with ALS maintain hearing, sight, touch, smell, and taste.[3]
Initial symptoms
[edit]The start of ALS may be so subtle that the symptoms are overlooked.[3] The earliest symptoms of ALS are muscle weakness or muscle atrophy, typically on one side of the body. Other presenting symptoms include trouble swallowing or breathing, cramping, or stiffness of affected muscles; muscle weakness affecting an arm or a leg; or slurred and nasal speech. The parts of the body affected by early symptoms of ALS depend on which motor neurons in the body are damaged first.[34]
In limb-onset ALS, the first symptoms are in the arms or the legs. If the legs are affected first, people may experience awkwardness, tripping, or stumbling when walking or running; this is often marked by walking with a "dropped foot" that drags gently on the ground. If the arms are affected first, they may experience difficulty with tasks requiring manual dexterity, such as buttoning a shirt, writing, or turning a key in a lock.[35]
In bulbar-onset ALS, the first symptoms are difficulty speaking or swallowing. Speech may become slurred, nasal in character, or quieter. There may be difficulty with swallowing and loss of tongue mobility. A smaller proportion of people experience "respiratory-onset" ALS, where the intercostal muscles that support breathing are affected first.[19]
Over time, people experience increasing difficulty moving, swallowing (dysphagia), and speaking or forming words (dysarthria). Symptoms of upper motor neuron involvement include tight and stiff muscles (spasticity) and exaggerated reflexes (hyperreflexia), including an overactive gag reflex.[24] While the disease does not cause pain directly, pain is a symptom experienced by most people with ALS caused by reduced mobility.[36] Symptoms of lower motor neuron degeneration include muscle weakness and atrophy, muscle cramps, and fleeting twitches of muscles that can be seen under the skin (fasciculations).[25]
Progression
[edit]Although the initial site of symptoms and subsequent rate of disability progression vary from person to person, the initially affected body region is usually the most affected over time, and symptoms usually spread to a neighbouring body region. For example, symptoms starting in one arm usually spread next to either the opposite arm or to the leg on the same side.[24] Bulbar-onset patients most typically get their next symptoms in their arms rather than legs, arm-onset patients typically spreads to the legs before the bulbar region, and leg-onset patients typically spread to the arms rather than the bulbar region.[37] Over time, regardless of where symptoms began, most people eventually lose the ability to walk or use their hands and arms independently. Less consistently, they may lose the ability to speak and to swallow food. It is the eventual development of weakness of the respiratory muscles, with the loss of ability to cough and to breathe without support, that is ultimately life-shortening in ALS.[5]
The rate of progression can be measured using the ALS Functional Rating Scale - Revised (ALSFRS-R), a 12-item instrument survey administered as a clinical interview or self-reported questionnaire that produces a score between 48 (normal function) and 0 (severe disability).[38] The ALSFRS-R is the most frequently used outcome measure in clinical trials[39] and is used by doctors to track disease progression.[40] Though the degree of variability is high and a small percentage of people have a much slower progression, on average people with ALS lose about 1 ALSFRS-R point per month.[41] Brief periods of stabilization ("plateaus") and even small reversals in ALSFRS-R score are not uncommon, due to the fact the tool is subjective, can be affected by medication, and different forms of compensation for changes in function.[42] However, it is rare (<1%) for these improvements to be large (i.e. greater than 4 ALSFRS-R points) or sustained (i.e. greater than 12 months).[42] A survey-based study among clinicians showed that they rated a 20% change in the slope of the ALSFRS-R as being clinically meaningful, which is the most common threshold used to determine whether a new treatment is working in clinical trials.[43]
Late stage disease management
[edit]Difficulties with chewing and swallowing make eating very difficult (dysphagia) and increase the risk of choking or of aspirating food into the lungs.[44] In later stages of the disorder, aspiration pneumonia can develop, and maintaining a healthy weight can become a significant problem that may require the insertion of a feeding tube.[44] As the diaphragm and intercostal muscles of the rib cage that support breathing weaken, measures of lung function such as vital capacity and inspiratory pressure diminish. In respiratory-onset ALS, this may occur before significant limb weakness is apparent. Individuals affected by the disorder may ultimately lose the ability to initiate and control all voluntary movement,[5] known as locked-in syndrome. Bladder and bowel function are usually spared, meaning urinary and fecal incontinence are uncommon, although trouble getting to a toilet can lead to difficulties. The extraocular muscles responsible for eye movement are usually spared,[45] meaning the use of eye tracking technology to support augmentative communication is often feasible, albeit slow, and needs may change over time.[46] Despite these challenges, many people in an advanced state of disease report satisfactory wellbeing and quality of life.[47]
Prognosis, staging, and survival
[edit]Although respiratory support using non-invasive ventilation can ease problems with breathing and prolong survival,[48] it does not affect the progression rate of ALS. Most people with ALS die between two and four years after the diagnosis.[5] Around 50% of people with ALS die within 30 months of their symptoms beginning, about 20% live between five and ten years,[19] and about 10% survive for 10 years or longer.[20]
The most common cause of death among people with ALS is respiratory failure, often accelerated by pneumonia.[19] Most ALS patients die at home after a period of worsening difficulty breathing, a decline in their nutritional status, or a rapid worsening of symptoms.[49] Sudden death or acute respiratory distress are uncommon.[50] Access to palliative care is recommended from an early stage to explore options, ensure psychosocial support for the patient and caregivers, and to discuss advance healthcare directives.[49]
As with cancer staging, ALS has staging systems numbered between 1 and 4 that are used for research purposes in clinical trials.[6] Two very similar staging systems emerged around a similar time, the King's staging system and Milano-Torino (MiToS) functional staging.[51]
| Stage 1 | Stage 2 | Stage 3 | Stage 4 | |
|---|---|---|---|---|
| Stage description | Symptom onset, involvement of the first region | 2A: Diagnosis 2B: Involvement of the second region | Involvement of the third region | 4A: Need for a feeding tube 4B: Need for non-invasive ventilation |
| Median time to stage | 13.5 months | 17.7 months | 23.3 months | 4A: 17.7 months 4B: 30.3 months |
| Stage 0 | Stage 1 | Stage 2 | Stage 3 | Stage 4 | Stage 5 | |
|---|---|---|---|---|---|---|
| Stage description | No loss of a functional domain | Loss of 1 domain | Loss of 2 domains | Loss of 3 domains | Loss of 4 domains | Death |
| Probability of death at each stage | 7% | 26% | 33% | 33% | 86% |
Providing individual patients with a precise prognosis is not currently possible, though research is underway to provide statistical models on the basis of prognostic factors including age at onset, progression rate, site of onset, and presence of frontotemporal dementia.[6] Those with a bulbar onset have a worse prognosis than limb-onset ALS; a population-based study found that bulbar-onset ALS patients had a median survival of 2.0 years and a 10-year survival rate of 3%, while limb-onset ALS patients had a median survival of 2.6 years and a 10-year survival rate of 13%.[52] Those with respiratory-onset ALS had a shorter median survival of 1.4 years and 0% survival at 10 years.[52] While astrophysicist Stephen Hawking lived for 55 more years following his diagnosis, his was an unusual case.[53]
Cognitive, emotional, and behavioral symptoms
[edit]Cognitive impairment or behavioral dysfunction is present in 30–50% of individuals with ALS,[54] and can appear more frequently in later stages of the disease.[55] Language dysfunction, executive dysfunction, and troubles with social cognition and verbal memory are the most commonly reported cognitive symptoms in ALS.[55] Cognitive impairment is found more frequently in patients with C9orf72 gene repeat expansions, bulbar onset, bulbar symptoms, family history of ALS, and/or a predominantly upper motor neuron phenotype.[56]
Emotional lability is a symptom in which patients cry, smile, yawn, or laugh, either in the absence of emotional stimuli, or when they are feeling the opposite emotion to that being expressed;[57] it is experienced by about half of ALS patients and is more common in those with bulbar-onset ALS.[5] While relatively benign relative to other symptoms, it can cause increased stigma and social isolation as people around the patient struggle to react appropriately to what can be frequent and inappropriate outbursts in public.[58]
In addition to mild changes in cognition that may only emerge during neuropsychological testing, around 10–15% of individuals have signs of frontotemporal dementia (FTD).[5] Repeating phrases or gestures, apathy, and loss of inhibition are the most frequently reported behavioral features of ALS.[59] ALS and FTD are now considered to be part of a common disease spectrum (ALS–FTD) because of genetic, clinical, and pathological similarities.[60] Genetically, repeat expansions in the C9orf72 gene account for about 40% of genetic ALS and 25% of genetic FTD.[61]
Cognitive and behavioral issues are associated with poorer prognosis as they may reduce adherence to medical advice, and deficits in empathy and social cognition which may increase caregiver burden.[62]
Cause
[edit]It is not known what causes sporadic ALS, hence it is described as an idiopathic disease.[19] Though its exact cause is unknown, genetic and environmental factors are thought to be of roughly equal importance.[12] The genetic factors are better understood than the environmental factors; no specific environmental factor has been definitively shown to cause ALS. A multi-step liability threshold model for ALS proposes that cellular damage accumulates over time due to genetic factors present at birth and exposure to environmental risks throughout life.[63] ALS can strike at any age, but its likelihood increases with age.[64] Most people who develop ALS are between the ages of 40 and 70, with an average age of 55 at the time of diagnosis.[65] ALS is 20% more common in men than women,[65] but this difference in sex distribution is no longer present in patients with onset after age 70.[64]
Genetics and genetic testing
[edit]While they appear identical clinically and pathologically,[66] ALS can be classified as being either familial or sporadic, depending on whether there is a known family history of the disease and/or whether an ALS-associated genetic mutation has been identified via genetic testing.[67] Familial ALS is thought to account for 10–15% of cases overall and can include monogenic, oligogenic, and polygenic modes of inheritance.[14]
There is considerable variation among clinicians on how to approach genetic testing in ALS, and only about half discuss the possibility of genetic inheritance with their patients, particularly if there is no discernible family history of the disease.[68] In the past, genetic counseling and testing was only offered to those with obviously familial ALS.[14] But it is increasingly recognized that cases of sporadic ALS may also be due to disease-causing de novo mutations in SOD1, or C9orf72,[69] an incomplete family history, or incomplete penetrance, meaning that a patient's ancestors carried the gene but did not express the disease in their lifetimes.[14] The lack of positive family history may be caused by lack of historical records, having a smaller family, older generations dying earlier of causes other than ALS, genetic non-paternity, and uncertainty over whether certain neuropsychiatric conditions (e.g. frontotemporal dementia, other forms of dementia, suicide, psychosis, schizophrenia) should be considered significant when determining a family history.[14] There have been calls in the research community to routinely counsel and test all diagnosed ALS patients for familial ALS,[70] particularly as there is now a licensed gene therapy (tofersen) specifically targeted to carriers of SOD-1 ALS. A shortage of genetic counselors and limited clinical capacity to see such at-risk individuals makes this challenging in practice, as does the unequal access to genetic testing around the world.[71]
More than 40 genes have been associated with ALS, of which four account for nearly half of familial cases, and around 5% of sporadic cases:[14] C9orf72 (40% of familial cases, 7% sporadic), SOD1 (12% of familial cases, 1–2% sporadic), FUS (4% of familial cases, 1% sporadic), and TARDBP (4% of familial cases, 1% sporadic), with the remaining genes mostly accounting for fewer than 1% of either familial or sporadic cases.[14] ALS genes identified to date explain the cause of about 70% of familial ALS and about 15% of sporadic ALS.[14] Overall, first-degree relatives of an individual with ALS have a ~1% risk of developing ALS themselves.[14]
Environmental and other factors
[edit]The multi-step hypothesis suggests the disease is caused by some interaction between an individual's genetic risk factors and their cumulative lifetime of exposures to environmental factors, termed their exposome.[14] The most consistent lifetime exposures associated with developing ALS (other than genetic mutations) include heavy metals (e.g. lead and mercury), chemicals (e.g. pesticides and solvents), electric shock, physical injury (including head injury), and smoking (in men more than women).[72] Overall these effects are small, with each exposure in isolation only increasing the likelihood of a very rare condition by a small amount. For instance an individual's lifetime risk of developing ALS might go from "1 in 400" without an exposure to between "1 in 300" and "1 in 200" if they were exposed to heavy metals.[72] A range of other exposures have weaker evidence supporting them and include participation in professional sports, having a lower body mass index, lower educational attainment, manual occupations, military service, exposure to Beta-N-methylamino-L-alanin (BMAA), and viral infections.[72]
Although some personality traits, such as openness,[73] agreeableness[74] and conscientiousness[74] appear remarkably common among patients with ALS, it remains open whether personality can increase susceptibility to ALS directly.[75] Instead, genetic factors giving rise to personality might simultaneously predispose people to developing ALS,[73] orthe above personality traits might underlie lifestyle choices which are in turn risk factors for ALS.[74]
Pathophysiology
[edit]Neuropathology
[edit]Upon examination at autopsy, features of the disease that can be seen with the naked eye include skeletal muscle atrophy, motor cortex atrophy, sclerosis of the corticospinal and corticobulbar tracts, thinning of the hypoglossal nerves (which control the tongue), and thinning of the anterior roots of the spinal cord.[10]
The defining feature of ALS is the death of both upper motor neurons (located in the motor cortex of the brain) and lower motor neurons (located in the brainstem and spinal cord).[76] In ALS with frontotemporal dementia, neurons throughout the frontal and temporal lobes of the brain die as well.[77] The pathological hallmark of ALS is the presence of inclusion bodies (abnormal aggregations of protein) known as Bunina bodies in the cytoplasm of motor neurons. In about 97% of people with ALS, the main component of the inclusion bodies is TDP-43 protein;[10] however, in those with SOD1 or FUS mutations, the main component of the inclusion bodies[78][79] is SOD1 protein or FUS protein, respectively.[24] Prion-like propagation of misfolded proteins from cell to cell may explain why ALS starts in one area and spreads to others.[24] The glymphatic system may also be involved in the pathogenesis of ALS.[80]
Biochemistry
[edit]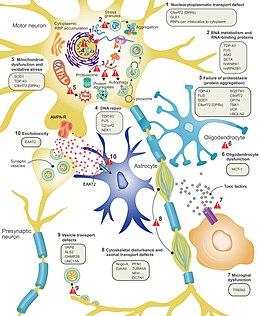
It is still not fully understood why neurons die in ALS, but this neurodegeneration is thought to involve many different cellular and molecular processes.[9] The genes known to be involved in ALS can be grouped into three general categories based on their normal function: protein degradation, the cytoskeleton, and RNA processing.[82] Mutant SOD1 protein forms intracellular aggregations that inhibit protein degradation. Cytoplasmic aggregations of wild-type (normal) SOD1 protein are common in sporadic ALS.[77] It is thought that misfolded mutant SOD1 can cause misfolding and aggregation of wild-type SOD1 in neighboring neurons in a prion-like manner.[10] Other protein degradation genes that can cause ALS when mutated include VCP, OPTN, TBK1, and SQSTM1. Three genes implicated in ALS that are important for maintaining the cytoskeleton[77] and for axonal transport[10] include DCTN1, PFN1, and TUBA4A.[77]
There are a number of ALS genes that encode for RNA-binding proteins. The first to be discovered was TDP-43 protein,[77] a nuclear protein that aggregates in the cytoplasm of motor neurons in almost all cases of ALS; however, mutations in TARDBP, the gene that codes for TDP-43, are a rare cause of ALS.[10] FUS codes for FUS, another RNA-binding protein with a similar function to TDP-43, which can cause ALS when mutated.[61] It is thought that mutations in TARDBP and FUS increase the binding affinity of the low-complexity domain, causing their respective proteins to aggregate in the cytoplasm.[83] Once these mutant RNA-binding proteins are misfolded and aggregated, they may be able to misfold normal proteins both within and between cells in a prion-like manner.[77] This also leads to decreased levels of RNA-binding protein in the nucleus, which may mean that their target RNA transcripts do not undergo normal processing. Other RNA metabolism genes associated with ALS include ANG, SETX, and MATR3.[10]
C9orf72 is the most commonly mutated gene in ALS and causes motor neuron death through a number of mechanisms.[77] The pathogenic mutation is a hexanucleotide repeat expansion (a series of six nucleotides repeated over and over);[84] people with up to 30 repeats are considered normal, while people with hundreds or thousands of repeats can have familial ALS, frontotemporal dementia, or sometimes sporadic ALS.[85] The three mechanisms of disease associated with these C9orf72 repeats are deposition of RNA transcripts in the nucleus, translation of the RNA into toxic dipeptide repeat proteins in the cytoplasm, and decreased levels of the normal C9orf72 protein.[77] Mitochondrial bioenergetic dysfunction leading to dysfunctional motor neuron axonal homeostasis (reduced axonal length and fast axonal transport of mitochondrial cargo) has been shown to occur in C9orf72-ALS using human induced pluripotent stem cell (iPSC) technologies coupled with CRISPR/Cas9 gene-editing, and human post-mortem spinal cord tissue examination.[86]
Excitotoxicity, or nerve cell death caused by high levels of intracellular calcium due to excessive stimulation by the excitatory neurotransmitter glutamate, is a mechanism thought to be common to all forms of ALS. Motor neurons are more sensitive to excitotoxicity than other types of neurons because they have a lower calcium-buffering capacity and a type of glutamate receptor (the AMPA receptor) that is more permeable to calcium. In ALS, there are decreased levels of excitatory amino acid transporter 2 (EAAT2), which is the main transporter that removes glutamate from the synapse; this leads to increased synaptic glutamate levels and excitotoxicity. Riluzole, a drug that modestly prolongs survival in ALS, inhibits glutamate release from pre-synaptic neurons; however, it is unclear if this mechanism is responsible for its therapeutic effect.[10]
Diagnosis
[edit]
No single test can provide a definite diagnosis of ALS.[3] Instead, the diagnosis of ALS is primarily made based on a physician's clinical assessment after ruling out other diseases.[3] Physicians often obtain the person's full medical history and conduct neurologic examinations at regular intervals to assess whether signs and symptoms such as muscle weakness, muscle atrophy, hyperreflexia, Babinski's sign, and spasticity are worsening.[3] A number of biomarkers are being studied for the condition, but as of 2023 are not in general medical use.[87]

Differential diagnosis
[edit]Because symptoms of ALS can be similar to those of a wide variety of other, more treatable diseases or disorders, appropriate tests must be conducted to exclude the possibility of other conditions. One of these tests is electromyography (EMG), a special recording technique that detects electrical activity in muscles. Certain EMG findings can support the diagnosis of ALS. Another common test measures nerve conduction velocity (NCV).[88] Specific abnormalities in the NCV results may suggest, for example, that the person has a form of peripheral neuropathy (damage to peripheral nerves) or myopathy (muscle disease) rather than ALS. While a magnetic resonance imaging (MRI) is often normal in people with early-stage ALS, it can reveal evidence of other problems that may be causing the symptoms, such as a spinal cord tumor, multiple sclerosis, a herniated disc in the neck, syringomyelia, or cervical spondylosis.[3]
Based on the person's symptoms and findings from the examination and from these tests, the physician may order tests on blood and urine samples to eliminate the possibility of other diseases, as well as routine laboratory tests. In some cases, for example, if a physician suspects the person may have a myopathy rather than ALS, a muscle biopsy may be performed.[3]
A number of infectious diseases can sometimes cause ALS-like symptoms,[3] including human immunodeficiency virus (HIV), human T-lymphotropic virus (HTLV), Lyme disease, and syphilis.[9] Neurological disorders such as multiple sclerosis, post-polio syndrome, multifocal motor neuropathy, CIDP, spinal muscular atrophy, and spinal and bulbar muscular atrophy can also mimic certain aspects of the disease and should be considered.[3]
ALS must be differentiated from the "ALS mimic syndromes", which are unrelated disorders that may have a similar presentation and clinical features to ALS or its variants.[89] Because the prognosis of ALS and closely related subtypes of motor neuron disease are generally poor, neurologists may carry out investigations to evaluate and exclude other diagnostic possibilities. Disorders of the neuromuscular junction, such as myasthenia gravis (MG) and Lambert–Eaton myasthenic syndrome, may also mimic ALS, although this rarely presents diagnostic difficulty over time.[90][91] Benign fasciculation syndrome and cramp fasciculation syndrome may also, occasionally, mimic some of the early symptoms of ALS. Nonetheless, the absence of other neurological features that develop inexorably with ALS means that, over time, the distinction will not present any difficulty to the experienced neurologist; where doubt remains, EMG may be helpful.[92]
Management
[edit]There is no cure for ALS.[9] Management focuses on treating symptoms and providing supportive care, with the goal of improving quality of life and prolonging survival.[9] This care is best provided by multidisciplinary teams of healthcare professionals; attending a multidisciplinary ALS clinic is associated with longer survival, fewer hospitalizations, and improved quality of life.[5]
Non-invasive ventilation (NIV) is the main treatment for respiratory failure in ALS.[10] In people with normal bulbar function, it prolongs survival by about seven months and improves quality of life. One study found that NIV is ineffective for people with poor bulbar function[93] while another suggested that it may provide a modest survival benefit.[9] Many people with ALS have difficulty tolerating NIV.[94] Invasive ventilation is an option for people with advanced ALS when NIV is not enough to manage their symptoms.[5] While invasive ventilation prolongs survival, disease progression and functional decline continue.[17] It may decrease the quality of life of people with ALS or their caregivers.[18][17] Invasive ventilation is more commonly used in Japan than in North America or Europe.[95]
Physical therapy can promote functional independence[96][97] through aerobic, range of motion, and stretching exercises.[98] Occupational therapy can assist with activities of daily living through adaptive equipment.[99] Speech therapy can assist people with ALS who have difficulty speaking.[97] Preventing weight loss and malnutrition in people with ALS improves both survival and quality of life.[9] Initially, difficulty swallowing (dysphagia) can be managed by dietary changes and swallowing techniques. A feeding tube should be considered if someone with ALS loses 5% or more of their body weight or if they cannot safely swallow food and water.[10] The feeding tube is usually inserted by percutaneous endoscopic gastrostomy (PEG). There is weak evidence that PEG tubes improve survival.[100] PEG insertion is usually performed with the intent of improving quality of life.[18]
Palliative care should begin shortly after someone is diagnosed with ALS.[101] Discussion of end-of-life issues gives people with ALS time to reflect on their preferences for end-of-life care and can help avoid unwanted interventions or procedures. Hospice care can improve symptom management at the end of life and increases the likelihood of a peaceful death.[18] In the final days of life, opioids can be used to treat pain and dyspnea, while benzodiazepines can be used to treat anxiety.[17]
Medications
[edit]Disease-slowing treatments
[edit]
Riluzole has been found to modestly prolong survival by about 2–3 months.[103][102] It may have a greater survival benefit for those with bulbar-onset ALS.[102] It may work by decreasing release of the excitatory neurotransmitter glutamate from pre-synaptic neurons.[10] The most common side effects are nausea and a lack of energy (asthenia).[102] People with ALS should begin treatment with riluzole as soon as possible following their diagnosis.[101] Riluzole is available as a tablet, liquid, or dissolvable oral film.[15]
Edaravone has been shown to modestly slow the decline in function in a small group of people with early-stage ALS.[104][105] It may work by protecting motor neurons from oxidative stress.[106] The most common side effects are bruising and gait disturbance.[105] Edaravone is available as an intravenous infusion or as an oral suspension.[107]
AMX0035 (Relyvrio) is a combination of sodium phenylbutyrate and taurursodiol, which was initially shown to prolong the survival of patients by an average of six months.[108] Relyvrio was withdrawn by the manufacturer in April 2024[109] following the completion of the Phase 3 PHOENIX trial[110] which did not show substantial benefit to ALS patients.
Tofersen (Qalsody) is an antisense oligonucleotide that was approved for medical use in the United States in April 2023, for the treatment of SOD1-associated ALS.[111] In a study of 108 patients with SOD1-associated ALS there was a non-significant trend towards a slowing of progression, as well as a significant reduction in neurofilament light chain,[112] a putative ALS biomarker thought to indicate neuronal damage.[113] A follow-up study and open-label extension suggested that earlier treatment initiation had a beneficial effect on slowing disease progression. Tofersen is available as an intrathecal injection into the lumbar cistern at the base of the spine.[111]
Symptomatic treatments
[edit]Other medications may be used to help reduce fatigue, ease muscle cramps, control spasticity, and reduce excess saliva and phlegm.[98] Gabapentin, pregabalin, and tricyclic antidepressants (e.g., amitriptyline) can be used for neuropathic pain, while nonsteroidal anti-inflammatory drugs (NSAIDs), acetaminophen, and opioids can be used for nociceptive pain.[36]
Депрессию можно лечить с помощью селективных ингибиторов обратного захвата серотонина (СИОЗС) или трициклических антидепрессантов. [10] while benzodiazepines can be used for anxiety.[5] There are no medications to treat cognitive impairment/frontotemporal dementia (FTD); however, SSRIs and antipsychotics can help treat some of the symptoms of FTD.[10] Baclofen and tizanidine are the most commonly used oral drugs for treating spasticity; an intrathecal baclofen pump can be used for severe spasticity.[10] Атропин , скополамин , амитриптилин или гликопирролат могут быть назначены, когда у людей с БАС возникают проблемы с глотанием слюны ( сиалорея ). [10]
Обзор 2017 года пришел к выводу, что мексилетин безопасен и эффективен для лечения судорог при БАС на основе рандомизированного контролируемого исследования 2016 года. [105]
Поддержка дыхания
[ редактировать ]Неинвазивная вентиляция
[ редактировать ]
Неинвазивная вентиляция легких (НИВ) является основным методом лечения дыхательной недостаточности при БАС. [10] и это был первый метод лечения, который улучшил как выживаемость, так и качество жизни. [5] При НИВЛ используется маска для лица или носа, подключенная к аппарату искусственной вентиляции легких, который обеспечивает периодическое положительное давление для поддержки дыхания. Постоянное положительное давление не рекомендуется людям с БАС, поскольку оно затрудняет дыхание. [17] Первоначально НИВЛ используется только ночью. [5] поскольку первым признаком дыхательной недостаточности является снижение газообмена ( гиповентиляция ) во время сна; Симптомы, связанные с ночной гиповентиляцией, включают прерывистый сон, беспокойство, утренние головные боли и дневную усталость. [94] По мере прогрессирования заболевания у людей с БАС развивается одышка в положении лежа, во время физической активности или разговора и, в конечном итоге, в состоянии покоя. [114] Другие симптомы включают плохую концентрацию, плохую память, спутанность сознания, инфекции дыхательных путей и слабый кашель. Дыхательная недостаточность является наиболее частой причиной смерти при БАС. [5]
Важно контролировать дыхательную функцию людей с БАС каждые три месяца, поскольку начало НИВЛ вскоре после появления респираторных симптомов связано с увеличением выживаемости. Это включает в себя опрос человека с БАС, есть ли у него какие-либо респираторные симптомы, и измерение его дыхательной функции. [5] Наиболее часто используемым методом измерения является форсированная жизненная емкость легких в вертикальном положении (ФЖЕЛ), но он является плохим индикатором ранней дыхательной недостаточности и не является хорошим выбором для людей с бульбарными симптомами, поскольку им трудно поддерживать плотное прилегание вокруг мундштука. Измерение ФЖЕЛ, когда человек лежит на спине (ФЖЕЛ лежа на спине), является более точным показателем слабости диафрагмы, чем ФЖЕЛ в вертикальном положении. [94] Нюхание давления на вдохе через нос (SNIP) — это быстрый и удобный тест силы диафрагмы, на который не влияет слабость бульбарных мышц. [17] Если у человека с БАС наблюдаются признаки и симптомы дыхательной недостаточности, ему следует пройти дневной анализ газов крови. [5] искать гипоксемию (низкий уровень кислорода в крови) и гиперкапнию (слишком много углекислого газа в крови). [17] Если анализ газов крови в дневное время в норме, им следует провести ночную пульсоксиметрию для выявления гипоксемии во время сна. [5]
Неинвазивная вентиляция продлевает выживаемость дольше, чем рилузол. [115] Рандомизированное контролируемое исследование 2006 года показало, что НИВЛ продлевает выживаемость примерно на 48 дней и улучшает качество жизни; однако также было обнаружено, что некоторые люди с БАС получают больше пользы от этого вмешательства, чем другие. Для людей с нормальной или умеренно нарушенной бульбарной функцией НИВЛ продлевает выживаемость примерно на семь месяцев и значительно улучшает качество жизни. Для людей с плохой бульбарной функцией НИВЛ не продлевает выживаемость и не улучшает качество жизни, хотя и улучшает некоторые симптомы, связанные со сном. [93] Несмотря на явные преимущества НИВЛ, около 25–30% всех людей с БАС не могут ее переносить, особенно те, у кого есть когнитивные нарушения или бульбарная дисфункция. [94] Результаты большого когортного исследования 2015 года показывают, что НИВЛ может продлить выживаемость у людей с бульбарной слабостью, поэтому НИВЛ следует предлагать всем людям с БАС, даже если вполне вероятно, что им будет трудно ее переносить. [9]
Инвазивная вентиляция
[ редактировать ]Инвазивная вентиляция минует нос и рот (верхние дыхательные пути) путем разреза трахеи ( трахеостома ) и введения трубки , подключенной к аппарату искусственной вентиляции легких. [17] Это вариант для людей с запущенным БАС, чьи респираторные симптомы плохо купируются, несмотря на постоянное использование НИВЛ. [5] Хотя инвазивная вентиляция легких продлевает выживаемость, особенно для лиц моложе 60 лет, она не лечит основной нейродегенеративный процесс. Человек с БАС будет продолжать терять двигательные функции, что затрудняет общение и иногда приводит к синдрому запертости , при котором он полностью парализован, за исключением глазных мышц. [17] Около половины людей с БАС, решивших пройти инвазивную вентиляцию легких, сообщают о снижении качества своей жизни. [18] но большинство все еще считает его удовлетворительным. Однако инвазивная вентиляция легких накладывает тяжелую нагрузку на лиц, осуществляющих уход, и может снизить качество их жизни. [17] Отношение к инвазивной вентиляции варьируется от страны к стране; около 30% людей с БАС в Японии выбирают инвазивную вентиляцию легких по сравнению с менее чем 5% в Северной Америке и Европе. [95]
Терапия
[ редактировать ]
Физиотерапия играет большую роль в реабилитации людей с БАС. В частности, физиотерапевты, эрготерапевты и логопеды могут ставить цели и продвигать преимущества для людей с БАС, замедляя потерю сил, поддерживая выносливость, ограничивая боль, улучшая речь и глотание, предотвращая осложнения и способствуя функциональной независимости. [96] [97]
Трудотерапия и специальное оборудование, такое как вспомогательные технологии, также могут повысить независимость и безопасность людей на протяжении всего периода БАС. [99] с низкой нагрузкой, Легкие аэробные упражнения такие как повседневная деятельность, ходьба, плавание и езда на велосипеде, могут укрепить непораженные мышцы, улучшить здоровье сердечно-сосудистой системы и помочь людям бороться с усталостью и депрессией. Диапазон движений и упражнения на растяжку могут помочь предотвратить болезненную спастичность и укорочение (контрактуру) мышц. [116] Физиотерапевты и эрготерапевты могут рекомендовать упражнения, которые обеспечивают эти преимущества без переутомления мышц, поскольку мышечное истощение может привести к ухудшению симптомов, связанных с БАС, вместо того, чтобы оказать помощь людям с БАС. [98] Они могут предложить такие устройства, как пандусы, подтяжки, ходунки, оборудование для ванных комнат (кресла для душа, стояки для унитазов и т. д.) и инвалидные коляски, которые помогают людям оставаться мобильными. Эрготерапевты могут предоставить или порекомендовать оборудование и приспособления, позволяющие людям с БАС сохранять как можно больше безопасности и независимости в повседневной жизни. [99] Поскольку дыхательная недостаточность является основной причиной смертности, физиотерапевты могут помочь улучшить респираторные исходы у людей с БАС, применяя легочную физиотерапию. Сюда входит тренировка мышц вдоха, тренировка набора объема легких и мануальная терапия кашля, направленная на увеличение силы дыхательных мышц, а также повышение выживаемости. [117]
Людям с БАС, испытывающим трудности с речью или глотанием, может быть полезна работа с логопедом . [97] Эти медицинские работники могут научить людей адаптивным стратегиям, таким как методы, которые помогут им говорить громче и четче. По мере прогрессирования БАС логопеды могут рекомендовать использование дополнительных и альтернативных средств связи , таких как усилители голоса, устройства генерации речи (или устройства голосовой связи) или низкотехнологичные методы связи, такие как наголовные лазерные указки, доски с алфавитом или сигналы да/нет. [97]
Питание
[ редактировать ]
Предотвращение потери веса и недоедания у людей с БАС улучшает как выживаемость, так и качество жизни. [9] Потеря веса при БАС часто вызвана атрофией мышц и увеличением затрат энергии в состоянии покоя. Потеря веса также может быть вторичной по отношению к уменьшению потребления пищи, поскольку дисфагия развивается примерно у 85% людей с БАС в какой-то момент течения заболевания. [17] Поэтому регулярная периодическая оценка веса и способности глотания у людей с БАС очень важна. [5] Дисфагию часто сначала лечат путем изменения диеты и модифицированной техники глотания. [10] Людям с БАС часто советуют избегать употребления сухих или жевательных продуктов и вместо этого есть мягкую, влажную и легко глотаемую пищу. [114] Переход на густые жидкости (например, фруктовый нектар или смузи) или добавление загустителей (к жидким жидкостям, таким как вода и кофе) также может помочь людям, испытывающим трудности с глотанием жидкости. Имеются предварительные доказательства того, что высококалорийные диеты могут предотвратить дальнейшую потерю веса и улучшить выживаемость. [105] но все еще необходимы дополнительные исследования.
Следует рассмотреть возможность использования зонда для кормления, если человек с БАС теряет 5% или более веса своего тела или если он не может безопасно глотать пищу и воду. [10] Это может быть гастростомическая трубка , при которой трубка вводится через стенку брюшной полости в желудок, или (реже) назогастральный зонд , при котором трубка вводится через нос и вниз по пищеводу в желудок. желудок. [17] Гастростомическая трубка больше подходит для длительного использования. [5] чем назогастральный зонд, который неудобен и может вызвать язву пищевода. [17] Питательную трубку обычно вводят с помощью чрескожной эндоскопической гастростомии (ЧЭГ). Хотя имеются слабые доказательства того, что использование ПЭГ-зондов улучшает выживаемость у людей с БАС, до сих пор не было проведено рандомизированных контролируемых исследований (РКИ), которые бы указали, имеет ли энтеральное зондовое питание преимущества по сравнению с продолжением кормления через рот. [100] Тем не менее, ПЭГ-трубки по-прежнему предлагаются с целью улучшения качества жизни человека. [18] поддерживая питание, уровень гидратации и прием лекарств. [5]
Уход в конце жизни
[ редактировать ]Паллиативная помощь , которая облегчает симптомы и улучшает качество жизни без лечения основного заболевания, должна начинаться вскоре после того, как у человека диагностирован БАС. [101] Раннее обсуждение проблем в конце жизни дает людям с БАС время подумать о своих предпочтениях в отношении ухода в конце жизни и может помочь избежать нежелательных вмешательств или процедур. [18] После того, как они будут полностью проинформированы обо всех аспектах различных мер по продлению жизни, они смогут заполнить предварительные инструкции , в которых будет указано их отношение к неинвазивной вентиляции, инвазивной вентиляции и зондам для кормления. [105] На поздних стадиях заболевания трудности с речью из-за мышечной слабости ( дизартрия ) и когнитивной дисфункции могут ухудшить их способность выражать свои пожелания относительно ухода. [10] Продолжительная неспособность выяснить предпочтения человека с БАС может привести к незапланированным и потенциально нежелательным экстренным вмешательствам, таким как инвазивная вентиляция легких. Если люди с БАС или члены их семей не хотят обсуждать вопросы конца жизни, может быть полезно использовать введение гастростомы или неинвазивной вентиляции как возможность поднять эту тему. [18]
Хосписная помощь , или паллиативная помощь в конце жизни, особенно важна при БАС, поскольку помогает оптимизировать лечение симптомов и увеличивает вероятность мирной смерти. [18] Неясно, когда именно начинается фаза конца жизни при БАС, но она связана со значительными трудностями при движении, общении и, в некоторых случаях, мышлении. [10] Хотя многие люди с БАС боятся удушья (удушья), [18] их можно заверить, что это происходит редко, менее чем в 1% случаев. [118] Большинство пациентов умирают дома, [18] а в последние дни жизни опиоиды можно использовать для лечения боли и одышки , а бензодиазепины — для лечения тревоги. [17]
Эпидемиология
[ редактировать ]БАС является наиболее распространенным заболеванием двигательных нейронов у взрослых и третьим по распространенности нейродегенеративным заболеванием. [61] после болезни Альцгеймера и болезни Паркинсона . [119] Во всем мире число людей, у которых ежегодно развивается БАС, оценивается в 1,9 человека на 100 000 в год, в то время как число людей, болеющих БАС в любой момент времени, оценивается примерно в 4,5 человека на 100 000. [120] В Европе количество новых случаев в год составляет около 2,6 человек на 100 000, а число заболевших — 7–9 человек на 100 000. [121] Пожизненный риск развития БАС составляет 1:350 для европейских мужчин и 1:400 для европейских женщин. Мужчины имеют более высокий риск главным образом потому, что БАС со спинальным началом чаще встречается у мужчин, чем у женщин. [63] Число людей с БАС в США в 2015 году составляло 5,2 человека на 100 000, причем оно было выше среди белых, мужчин и людей старше 60 лет. [122] Число новых случаев составляет около 0,8 человека на 100 000 в год в Восточной Азии и около 0,7 человека на 100 000 в год в Южной Азии. Около 80% эпидемиологических исследований БАС было проведено в Европе и США, в основном среди людей североевропейского происхождения. [10] Недостаточно информации для определения распространенности БАС в большей части мира, включая Африку, некоторые части Азии, Индию, Россию и Южную Америку. [63] В западной части Тихого океана есть несколько географических кластеров, где распространенность БАС, как сообщается, в 50–100 раз выше, чем в остальном мире, включая Гуам , полуостров Кии в Японии и западную часть Новой Гвинеи . Заболеваемость в этих районах снизилась с 1960-х годов; [1] причина остается неизвестной. [63]

Люди всех рас и этнических групп могут страдать от БАС. [122] но это чаще встречается у белых, чем у африканцев, азиатов или латиноамериканцев. [123] В США в 2015 году распространенность БАС среди белых составила 5,4 человека на 100 000, а среди чернокожих — 2,3 человека на 100 000. На Среднем Западе наблюдалась самая высокая распространенность из четырех регионов переписи населения США - 5,5 человек на 100 000, за ним следовали Северо-Восток (5,1), Юг (4,7) и Запад (4,4). На Среднем Западе и Северо-Востоке, вероятно, наблюдалась более высокая распространенность БАС, поскольку там более высокая доля белого населения, чем на Юге и Западе. [122] Этнически смешанные группы населения могут подвергаться меньшему риску развития БАС; Исследование, проведенное на Кубе, показало, что люди смешанного происхождения реже умирают от БАС, чем белые или чернокожие. [124] Существуют также различия в генетике БАС между разными этническими группами; наиболее распространенным геном БАС в Европе является C9orf72 , за ним следуют SOD1 , TARDBP и FUS , тогда как наиболее распространенным геном БАС в Азии является SOD1 , за которым следуют FUS , C9orf72 и TARDBP . [125]
БАС может поражать людей в любом возрасте, [54] но пик заболеваемости приходится на период от 50 до 75 лет. [9] и резко снижается после 80 лет. [19] Причина снижения заболеваемости среди пожилых людей неясна. Одна из идей заключается в том, что люди, доживающие до 80-летнего возраста, могут быть генетически не предрасположены к развитию БАС; альтернативно, БАС у пожилых людей может остаться невыявленным из-за сопутствующих заболеваний (других заболеваний), трудностей с посещением невролога или быстрой смерти от агрессивной формы БАС. [124] В США в 2015 году самая низкая распространенность наблюдалась в возрастной группе 18–39 лет, а самая высокая распространенность – в возрастной группе 70–79 лет. [122] Спорадический БАС обычно начинается в возрасте от 58 до 63 лет, тогда как генетический БАС начинается раньше, обычно в возрасте от 47 до 52 лет. [19] По прогнозам, число случаев БАС во всем мире увеличится с 222 801 в 2015 году до 376 674 в 2040 году, то есть на 69%. Во многом это будет связано со старением населения мира, особенно в развивающихся странах. [123]
История
[ редактировать ]

Описания болезни датируются как минимум 1824 годом Чарльзом Беллом . [21] В 1850 году Франсуа-Амилькар Аран первым описал заболевание, которое он назвал «прогрессирующей мышечной атрофией», формой БАС, при которой поражаются только нижние мотонейроны. [127] В 1869 году связь между симптомами и лежащими в их основе неврологическими проблемами была впервые описана Жаном-Мартеном Шарко , который впервые ввел термин «боковой амиотрофический склероз» в своей статье 1874 года. [21] Синдром цепной руки, региональный вариант БАС, был впервые описан Альфредом Вульпианом в 1886 году. Синдром цепной ноги, еще один региональный вариант БАС, был впервые описан Пьером Мари и его учеником Патрикиосом в 1918 году. [128]
Диагностические критерии
[ редактировать ]В 1950-х годах электродиагностическое тестирование (ЭМГ) и тестирование скорости нервной проводимости для оценки клинически подозреваемого БАС стали использовать (NCV). В 1969 году Эдвард Х. Ламберт опубликовал первые диагностические критерии БАС с помощью ЭМГ/NCS, состоящие из четырех результатов, которые, по его мнению, убедительно подтверждают диагноз. [129] С тех пор был разработан ряд диагностических критериев, которые в основном используются в исследовательских целях для определения критериев включения/исключения, а также для стратификации пациентов для анализа в исследованиях. Критерии диагностики БАС включают исследование «Эль Эскориал» в 1994 г., [130] пересмотрено в 1998 году. [131] В 2006 году критерии «Аваджи» предложили использовать тесты ЭМГ и NCV для более ранней диагностики БАС. [132] и совсем недавно критерии «Золотого Берега» в 2019 году. [133]
Имя
[ редактировать ]Амиотрофический происходит от греческого : а- означает «нет», мио- (от mûs ) означает «мышца», а трофе означает «питание». Следовательно, амиотрофия означает «недоедание мышц». [134] или истощение мышечной ткани. [135] Латеральный идентифицирует места в спинном мозге пораженных мотонейронов. Склероз означает «рубцевание» или «затвердевание» и означает гибель мотонейронов спинного мозга. [134]
БАС иногда называют болезнью Шарко (не путать с болезнью Шарко-Мари-Тута или болезнью суставов Шарко ), поскольку Жан-Мартен Шарко был первым, кто связал клинические симптомы с патологией, наблюдаемой при аутопсии. [136] Британский невролог Рассел Брейн ввел термин «болезнь двигательных нейронов» в 1933 году, чтобы отразить свое убеждение в том, что БАС, прогрессирующий бульбарный паралич и прогрессирующая мышечная атрофия — это разные формы одного и того же заболевания. [137] В некоторых странах, особенно в США, БАС называют болезнью Лу Герига. [126] честь американского бейсболиста в Лу Герига , которому в 1939 году был поставлен диагноз БАС. [138]
В Соединенных Штатах и континентальной Европе термин БАС (а также болезнь Лу Герига в США) относится ко всем формам заболевания, включая «классический» БАС, прогрессирующий бульбарный паралич , прогрессирующую мышечную атрофию и первичный боковой склероз . [139] [33] В Соединенном Королевстве и Австралии термин « болезнь двигательных нейронов» относится ко всем формам заболевания, тогда как БАС относится только к «классическому» БАС, то есть к форме с поражением как верхних, так и нижних двигательных нейронов. [139]
Общество и культура
[ редактировать ]Помимо бейсболиста Лу Герига и физика-теоретика Стивена Хокинга (который прожил значительно дольше, чем любой другой известный человек с этим заболеванием), ряд других известных людей страдали или страдали БАС. [65] О пациентах с этим заболеванием написано несколько книг и сняты фильмы. Американский профессор социологии и пациент БАС Морри Шварц стал героем мемуаров «Вторники с Морри» и одноименного фильма , а Стивен Хокинг стал героем получившего признание критиков биографического фильма «Теория всего» .
В августе 2014 года конкурс Ice Bucket Challenge по сбору денег на исследования БАС стал вирусным в Интернете. [140] Участники снимали, как наполняют ведро ледяной водой и выливают ее на себя; затем они назначили других людей сделать то же самое. Многие участники сделали пожертвования на исследования БАС в Ассоциации БАС , Институте развития терапии БАС , Канадском обществе БАС или Ассоциации заболеваний двигательных нейронов в Великобритании. [141]
Ссылки
[ редактировать ]![]() В эту статью включен текст из этого источника, который находится в свободном доступе : «Информационный бюллетень по боковому амиотрофическому склерозу» . Архивировано из оригинала 18 ноября 2004 года.
В эту статью включен текст из этого источника, который находится в свободном доступе : «Информационный бюллетень по боковому амиотрофическому склерозу» . Архивировано из оригинала 18 ноября 2004 года.
- ^ Перейти обратно: а б Виджесекера LC, Ли П.Н. (февраль 2009 г.). «Боковой амиотрофический склероз» . Сиротский журнал редких заболеваний . 4 (4): 3. дои : 10.1186/1750-1172-4-3 . ПМЦ 2656493 . ПМИД 19192301 .
- ^ Перейти обратно: а б с д и ж г час я Масрори П., Ван Дамм П. (октябрь 2020 г.). «Боковой амиотрофический склероз: клинический обзор» . Европейский журнал неврологии . 27 (10): 1918–1929. дои : 10.1111/ene.14393 . ПМЦ 7540334 . ПМИД 32526057 .
- ^ Перейти обратно: а б с д и ж г час я дж к л м н тот п д р «Информационный бюллетень о боковом амиотрофическом склерозе (БАС)» . Национальный институт неврологических расстройств и инсульта. Архивировано из оригинала 5 января 2017 года . Проверено 22 октября 2020 г.
- ^ Кван Дж., Вуллаганти М. (сентябрь 2022 г.). «Имитирует боковой амиотрофический склероз». Мышцы и нервы . 66 (3): 240–252. дои : 10.1002/mus.27567 . ПМИД 35607838 . S2CID 249014375 .
- ^ Перейти обратно: а б с д и ж г час я дж к л м н тот п д р с т Хобсон Е.В., Макдермотт С.Дж. (сентябрь 2016 г.). «Поддерживающее и симптоматическое лечение бокового амиотрофического склероза» (PDF) . Обзоры природы. Неврология . 12 (9): 526–538. дои : 10.1038/nrneurol.2016.111 . ПМИД 27514291 . S2CID 8547381 . Архивировано (PDF) из оригинала 1 декабря 2020 года . Проверено 20 декабря 2019 г.
- ^ Перейти обратно: а б с д и ж г час Гутман С.А., Хардиман О., Аль-Чалаби А., Чио А., Савельев М.Г., Кирнан М.К., Фельдман Э.Л. (май 2022 г.). «Последние достижения в диагностике и прогнозировании бокового амиотрофического склероза» . «Ланцет». Неврология . 21 (5): 480–493. дои : 10.1016/S1474-4422(21)00465-8 . ПМЦ 9513753 . ПМИД 35334233 .
- ^ Райан М., Хеверин М., Маклафлин Р.Л., Хардиман О. (ноябрь 2019 г.). «Пожизненный риск и наследственность бокового амиотрофического склероза» . JAMA Неврология . 76 (11): 1367–1374. дои : 10.1001/jamaneurol.2019.2044 . ПМК 6646974 . ПМИД 31329211 .
- ^ «Информационный бюллетень о заболеваниях двигательных нейронов» . www.ninds.nih.gov . Национальный институт неврологических расстройств и инсульта. Архивировано из оригинала 10 октября 2020 года . Проверено 27 октября 2020 г.
- ^ Перейти обратно: а б с д и ж г час я дж к л м н тот п ван Эс М.А., Хардиман О., Чио А., Аль-Чалаби А., Пастеркамп Р.Дж., Вельдинк Дж.Х., ван ден Берг Л.Х. (ноябрь 2017 г.). «Боковой амиотрофический склероз». Ланцет . 390 (10107): 2084–2098. дои : 10.1016/S0140-6736(17)31287-4 . ПМИД 28552366 . S2CID 24483077 .
- ^ Перейти обратно: а б с д и ж г час я дж к л м н тот п д р с т в v Хардиман О., Аль-Чалаби А., Чио А., Корр Э.М., Логроскино Г., Робберехт В. и др. (октябрь 2017 г.). «Боковой амиотрофический склероз» (PDF) . Обзоры природы. Праймеры по болезням . 3 (17071): 17071. doi : 10.1038/nrdp.2017.71 . ПМИД 28980624 . S2CID 1002680 . Архивировано (PDF) из оригинала 1 декабря 2020 года . Проверено 20 декабря 2019 г.
- ^ «Понимание БАС» . Ассоциация БАС. Архивировано из оригинала 26 октября 2020 года . Проверено 28 октября 2020 г.
- ^ Перейти обратно: а б Винго Т.С., Катлер DJ, Яраб Н., Келли СМ, Гласс ДжейДи (2011). «Наследственность бокового амиотрофического склероза в клинически подтвержденном реестре исследований США» . ПЛОС ОДИН . 6 (11): e27985. Бибкод : 2011PLoSO...627985W . дои : 10.1371/journal.pone.0027985 . ПМЦ 3222666 . ПМИД 22132186 .
- ^ «Боковой амиотрофический склероз» . МедлайнПлюс Генетика . Проверено 7 августа 2023 г.
- ^ Перейти обратно: а б с д и ж г час я дж Гутман С.А., Хардиман О., Аль-Чалаби А., Чио А., Савельев М.Г., Кирнан М.К., Фельдман Э.Л. (май 2022 г.). «Новые знания о сложной генетике и патофизиологии бокового амиотрофического склероза» . «Ланцет». Неврология . 21 (5): 465–479. дои : 10.1016/S1474-4422(21)00414-2 . ПМЦ 9513754 . ПМИД 35334234 .
- ^ Перейти обратно: а б «Одобренные FDA препараты для лечения БАС» . Ассоциация БАС . Архивировано из оригинала 25 апреля 2023 года . Проверено 25 апреля 2023 г.
- ^ Хо Ю.А. (сентябрь 2022 г.). «Фенилбутират натрия и урсодококсиколтаурин: первое одобрение» . Препараты ЦНС . 36 (9): 1007–1013. дои : 10.1007/s40263-022-00945-x . ПМИД 35907175 . S2CID 251162676 .
- ^ Перейти обратно: а б с д и ж г час я дж к л м н Сориани М.Х., Деснуэль С. (май 2017 г.). «Организация медицинской помощи при боковом амиотрофическом склерозе». Ревю Неврологии . 173 (5): 288–299. дои : 10.1016/j.neurol.2017.03.031 . ПМИД 28461024 .
- ^ Перейти обратно: а б с д и ж г час я дж к Коннолли С., Гэлвин М., Хардиман О. (апрель 2015 г.). «Ведение пациентов в конце жизни с боковым амиотрофическим склерозом». «Ланцет». Неврология . 14 (4): 435–442. дои : 10.1016/S1474-4422(14)70221-2 . ПМИД 25728958 . S2CID 34109901 .
- ^ Перейти обратно: а б с д и ж г час я Кирнан М.К., Вучич С., Чеа Б.С., Тернер М.Р., Эйзен А., Хардиман О. и др. (март 2011 г.). «Боковой амиотрофический склероз» . Ланцет . 377 (9769): 942–955. дои : 10.1016/s0140-6736(10)61156-7 . ПМИД 21296405 .
- ^ Перейти обратно: а б Пупилло Э., Мессина П., Логроскино Г., Беги Э. (февраль 2014 г.). «Долгосрочная выживаемость при боковом амиотрофическом склерозе: популяционное исследование». Анналы неврологии . 75 (2): 287–297. дои : 10.1002/ana.24096 . ПМИД 24382602 . S2CID 205345019 .
- ^ Перейти обратно: а б с д и Роуленд LP (март 2001 г.). «Как боковой амиотрофический склероз получил свое название: клинико-патологический гений Жан-Мартена Шарко». Архив неврологии . 58 (3): 512–515. дои : 10.1001/archneur.58.3.512 . ПМИД 11255459 .
- ^ «8B60 Болезнь двигательных нейронов» . МКБ-11 по статистике смертности и заболеваемости . Всемирная организация здравоохранения. Архивировано из оригинала 1 августа 2018 года . Проверено 24 января 2019 г.
- ^ ван Эненнаам Р.М., Коппенол Л.С., Круитхоф В.Дж., Круитваген-ван Реенен Э.Т., Питерс С., ван Эс М.А. и др. (ноябрь 2021 г.). «Обсуждение персонализированного прогноза дает пациентам с боковым амиотрофическим склерозом возможность восстановить контроль над своим будущим: качественное исследование» . Науки о мозге . 11 (12): 1597. doi : 10.3390/brainsci11121597 . ПМЦ 8699408 . ПМИД 34942899 .
- ^ Перейти обратно: а б с д и ж г час Град Л.И., Руло Г.А., Равиц Дж., Кэшман Н.Р. (август 2017 г.). «Клинический спектр бокового амиотрофического склероза (АЛС)» . Перспективы Колд-Спринг-Харбора в медицине . 7 (8): а024117. doi : 10.1101/cshperspect.a024117 . ПМК 5538408 . ПМИД 28003278 .
- ^ Перейти обратно: а б Арора Р.Д., Хан Ю.С. (2023). «Болезнь двигательных нейронов» . СтатПерлс . Остров сокровищ (Флорида): StatPearls Publishing. ПМИД 32809609 . Проверено 27 августа 2023 г.
- ^ Готье Дж., Вершуерен А., Моннье А., Аттариан С., Салорт-Кампана Э., Пуже Дж. (август 2010 г.). «АЛС с респираторным началом: клинические особенности и влияние неинвазивной вентиляции легких на прогноз». Боковой амиотрофический склероз . 11 (4): 379–382. дои : 10.3109/17482960903426543 . ПМИД 20001486 . S2CID 27672209 .
- ^ Перейти обратно: а б с Свиннен Б., Робберехт В. (ноябрь 2014 г.). «Фенотипическая изменчивость бокового амиотрофического склероза» . Обзоры природы. Неврология . 10 (11): 661–670. дои : 10.1038/nrneurol.2014.184 . ПМИД 25311585 . S2CID 205516010 . Архивировано из оригинала 31 декабря 2018 года . Проверено 22 августа 2018 г.
- ^ Перейти обратно: а б с Аль-Чалаби А., Хардиман О., Кирнан М.К., Чио А., Рикс-Брукс Б., ван ден Берг Л.Х. (октябрь 2016 г.). «Боковой амиотрофический склероз: движение к новой системе классификации». «Ланцет». Неврология . 15 (11): 1182–1194. дои : 10.1016/S1474-4422(16)30199-5 . hdl : 2318/1636249 . ПМИД 27647646 . S2CID 45285510 .
- ^ Джавдат О, Стэтленд Дж. М., Барон Р. Дж., Кац Дж. С., Димачки М. М. (ноябрь 2015 г.). «Региональные варианты бокового амиотрофического склероза (амиотрофическая диплегия плечевого сустава, амиотрофическая диплегия ног и изолированный бульбарный боковой амиотрофический склероз)» . Неврологические клиники . 33 (4): 775–785. дои : 10.1016/j.ncl.2015.07.003 . ПМЦ 4629514 . ПМИД 26515621 .
- ^ Чжан Х, Чен Л, Тянь Дж, Фань Д (октябрь 2021 г.). «Длительность прогрессирования заболевания помогает идентифицировать изолированный бульбарный паралич при боковом амиотрофическом склерозе» . БМК Неврология . 21 (1): 405. doi : 10.1186/s12883-021-02438-8 . ПМЦ 8532334 . ПМИД 34686150 .
- ^ Леки Т., Грюнсейх С. (ноябрь 2021 г.). «Ювенильный боковой амиотрофический склероз: обзор» . Гены . 12 (12): 1935. doi : 10.3390/genes12121935 . ПМК 8701111 . ПМИД 34946884 .
- ^ Теох Х.Л., Кэри К., Сампайо Х., Моват Д., Росчиоли Т., Фаррар М. (2017). «Наследственные педиатрические заболевания двигательных нейронов: помимо спинальной мышечной атрофии» . Нейронная пластичность . 2017 : 6509493. doi : 10.1155/2017/6509493 . ПМЦ 5467325 . ПМИД 28634552 .
- ^ Перейти обратно: а б Тард С., Дефевр Л., Моро С., Девос Д., Данель-Брюно В. (май 2017 г.). «Клинические особенности бокового амиотрофического склероза и их прогностическое значение». Ревю Неврологии . 173 (5): 263–272. дои : 10.1016/j.neurol.2017.03.029 . ПМИД 28477850 .
- ^ Равитс Дж., Аппель С., Балох Р.Х., Барон Р., Брукс Б.Р., Элман Л. и др. (май 2013 г.). «Расшифровка бокового амиотрофического склероза: что фенотип, невропатология и генетика говорят нам о патогенезе» . Боковой амиотрофический склероз и лобно-височная дегенерация . 14 (Приложение 1): 5–18. дои : 10.3109/21678421.2013.778548 . ПМЦ 3779649 . ПМИД 23678876 .
- ^ «Болезнь двигательных нейронов» . Великобритания: Национальная служба здравоохранения. 15 января 2018 года. Архивировано из оригинала 29 декабря 2014 года . Проверено 24 октября 2020 г.
- ^ Перейти обратно: а б Чио А., Мора Г., Лаурия Г. (февраль 2017 г.). «Боль при боковом амиотрофическом склерозе». «Ланцет». Неврология . 16 (2): 144–157. arXiv : 1607.02870 . дои : 10.1016/S1474-4422(16)30358-1 . ПМИД 27964824 . S2CID 38905437 .
- ^ Громичо М., Фигейрал М., Уйсал Х., Гросскройц Дж., Кузьма-Козакевич М., Пинто С. и др. (июль 2020 г.). «Распространение при БАС: относительное влияние поражения верхних и нижних мотонейронов» . Анналы клинической и трансляционной неврологии . 7 (7): 1181–1192. дои : 10.1002/acn3.51098 . ПМЦ 7359118 . ПМИД 32558369 .
- ^ Седарбаум Дж. М., Стамблер Н., Мальта Э., Фуллер С., Хилт Д., Турмонд Б., Наканиши А. (октябрь 1999 г.). «ALSFRS-R: пересмотренная функциональная рейтинговая шкала БАС, которая включает оценку дыхательной функции. Группа исследования БАС BDNF (Фаза III)». Журнал неврологических наук . 169 (1–2): 13–21. дои : 10.1016/s0022-510x(99)00210-5 . ПМИД 10540002 . S2CID 7057926 .
- ^ Вонг С., Ставру М., Эллиотт Э., Грегори Дж.М., Ли Н., Пинто А.А. и др. (2021). «Клинические исследования бокового амиотрофического склероза: систематический обзор и перспективы» . Мозговые коммуникации . 3 (4): fcab242. doi : 10.1093/braincomms/fcab242 . ПМЦ 8659356 . ПМИД 34901853 .
- ^ Кримерс Х., Группа Х., Ноллет Ф., ван ден Берг Л.Х., Билен А. (июнь 2015 г.). «Прогностические факторы течения функционального состояния больных БАС: систематический обзор». Журнал неврологии . 262 (6): 1407–1423. дои : 10.1007/s00415-014-7564-8 . ПМИД 25385051 . S2CID 31734765 .
- ^ Атасси Н., Берри Дж., Шуй А., Зак Н., Шерман А., Синани Э. и др. (ноябрь 2014 г.). «База данных PRO-ACT: дизайн, первоначальный анализ и прогностические функции» . Неврология . 83 (19): 1719–1725. дои : 10.1212/WNL.0000000000000951 . ПМЦ 4239834 . ПМИД 25298304 .
- ^ Перейти обратно: а б Бедлак Р.С., Воан Т., Уикс П., Хейвуд Дж., Синани Э., Сельсов Р. и др. (март 2016 г.). «Насколько распространены плато и развороты БАС?» . Неврология . 86 (9): 808–812. дои : 10.1212/WNL.0000000000002251 . ПМЦ 4793781 . ПМИД 26658909 .
- ^ Кастрильо-Вигера С., Грассо Д.Л., Симпсон Э., Шефнер Дж., Кудкович М.Е. (2010). «Клиническое значение изменения снижения ALSFRS-R». Боковой амиотрофический склероз (журнальная статья). 11 (1–2): 178–180. дои : 10.3109/17482960903093710 . ПМИД 19634063 . S2CID 207619689 .
- ^ Перейти обратно: а б Юнусова Ю., Пахарь Е.К., Грин Дж.Р., Барнетт С., Беде П. (2019). «Клинические меры бульбарной дисфункции при БАС» . Границы в неврологии . 10 :106. doi : 10.3389/fneur.2019.00106 . ПМК 6389633 . ПМИД 30837936 .
- ^ Луи А.Дж., Был Н.Н. (июнь 2009 г.). «Систематический обзор влияния упражнений средней интенсивности на функцию и прогрессирование заболевания при боковом амиотрофическом склерозе» . Журнал неврологической физиотерапии . 33 (2): 68–87. дои : 10.1097/NPT.0b013e31819912d0 . ПМИД 19556916 . S2CID 7650356 .
- ^ Бойкельман Д., Фагер С., Норднесс А. (2011). «Коммуникационная поддержка людей с БАС» . Международное исследование неврологии . 2011 : 714693. doi : 10.1155/2011/714693 . ПМК 3096454 . ПМИД 21603029 .
- ^ Кузьма-Козакевич М., Андерсен П.М., Цецверска К., Васкес С., Хельчик О., Лузе М. и др. (сентябрь 2019 г.). «Обсервационное исследование качества жизни и предпочтений поддерживать жизнь в запертом состоянии» . Неврология . 93 (10): е938–е945. дои : 10.1212/WNL.0000000000008064 . ПМЦ 6745736 . ПМИД 31391247 .
- ^ О'Брайен Д., Ставрулакис Т., Бакстер С., Норман П., Бьянки С., Эллиот М. и др. (сентябрь 2019 г.). «Оптимизация неинвазивной вентиляции при боковом амиотрофическом склерозе: систематический обзор» . Европейский респираторный журнал . 54 (3): 1900261. doi : 10.1183/13993003.00261-2019 . ПМИД 31273038 . S2CID 195805546 .
- ^ Перейти обратно: а б Беде П., Оливер Д., Стодарт Дж., ван ден Берг Л., Симмонс З., О. Браннагайн Д. и др. (апрель 2011 г.). «Паллиативная помощь при боковом амиотрофическом склерозе: обзор текущих международных рекомендаций и инициатив» . Журнал неврологии, нейрохирургии и психиатрии . 82 (4): 413–418. дои : 10.1136/jnnp.2010.232637 . hdl : 2262/59035 . ПМИД 21297150 . S2CID 7043837 . Архивировано из оригинала 28 мая 2023 года . Проверено 30 апреля 2023 г.
- ^ Корсиа П., Прадат П.Ф., Салачас Ф., Брюнето Дж., Форестье Н., Сейлен Д. и др. (1 января 2008 г.). «Причины смерти в серии вскрытий больных БАС». Боковой амиотрофический склероз . 9 (1): 59–62. дои : 10.1080/17482960701656940 . ПМИД 17924236 . S2CID 40367873 .
- ^ Фанг Т., Аль Хлейфат А., Шталь Д.Р., Лазо Ла Торре С., Мерфи С., Янг С. и др. (май 2017 г.). «Сравнение систем постановки Кинга и MiToS при БАС» . Боковой амиотрофический склероз и лобно-височная дегенерация . 18 (3–4): 227–232. дои : 10.1080/21678421.2016.1265565 . ПМЦ 5425622 . ПМИД 28054828 .
- ^ Перейти обратно: а б Чио А., Кальво А., Молья С., Мадзини Л., Мора Г. (июль 2011 г.). «Фенотипическая гетерогенность бокового амиотрофического склероза: популяционное исследование» (PDF) . Журнал неврологии, нейрохирургии и психиатрии . 82 (7): 740–746. дои : 10.1136/jnnp.2010.235952 . ПМИД 21402743 . S2CID 13416164 . Архивировано (PDF) из оригинала 24 ноября 2022 года . Проверено 4 августа 2022 г.
- ^ Ландау Э. (20 сентября 2009 г.). «Стивен Хокинг служит образцом для подражания для пациентов с БАС» . Си-Эн-Эн. Архивировано из оригинала 15 августа 2016 года.
- ^ Перейти обратно: а б Мартин С., Аль Хлейфат А., Аль-Чалаби А. (2017). «Что вызывает боковой амиотрофический склероз?» . F1000Исследования . 6 : 371. дои : 10.12688/f1000research.10476.1 . ПМЦ 5373425 . ПМИД 28408982 .
- ^ Перейти обратно: а б Крокфорд С., Ньютон Дж., Лонерган К., Чивера Т., Бут Т., Чандран С. и др. (октябрь 2018 г.). «Специфические для БАС когнитивные и поведенческие изменения, связанные с прогрессированием стадии заболевания при БАС» . Неврология . 91 (15): е1370–е1380. дои : 10.1212/WNL.0000000000006317 . ПМК 6177274 . ПМИД 30209236 .
- ^ Ян Т, Хоу Ю, Ли С, Цао Б, Ченг Ю, Вэй Ц и др. (июль 2021 г.). «Факторы риска когнитивных нарушений при боковом амиотрофическом склерозе: систематический обзор и метаанализ». Журнал неврологии, нейрохирургии и психиатрии . 92 (7): 688–693. дои : 10.1136/jnnp-2020-325701 . ПМИД 33563800 . S2CID 231858696 .
- ^ Уикс П. (июль 2007 г.). «Чрезмерное зевание часто встречается при бульбарной форме БАС». Acta Psychiatrica Scandinavica . 116 (1): 76, ответ автора 76–76, ответ автора 77. doi : 10.1111/j.1600-0447.2007.01025.x . ПМИД 17559605 . S2CID 12807996 .
- ^ Сове WM (декабрь 2016 г.). «Распознавание и лечение псевдобульбарного аффекта». Спектры ЦНС . 21 (С1): 34–44. дои : 10.1017/S1092852916000791 . ПМИД 28044945 . S2CID 21066800 .
- ^ Раапхорст Дж., Бильдман Э., Де Виссер М., Де Хаан Р.Дж., Шманд Б. (октябрь 2012 г.). «Систематический обзор поведенческих изменений при заболевании двигательных нейронов». Боковой амиотрофический склероз . 13 (6): 493–501. дои : 10.3109/17482968.2012.656652 . ПМИД 22424127 . S2CID 22224140 .
- ^ Куратье П., Корсия П., Лотретт Дж., Николь М., Марин Б. (май 2017 г.). «БАС и лобно-височная деменция принадлежат к общему спектру заболеваний». Ревю Неврологии . 173 (5): 273–279. дои : 10.1016/j.neurol.2017.04.001 . ПМИД 28449882 .
- ^ Перейти обратно: а б с Рентон А.Э., Чио А., Трейнор Б.Дж. (январь 2014 г.). «Состояние генетики бокового амиотрофического склероза» . Природная неврология . 17 (1): 17–23. дои : 10.1038/nn.3584 . hdl : 2318/156177 . ПМЦ 4544832 . ПМИД 24369373 .
- ^ Бильдман Э., Раапхорст Дж., Кляйн Твеннаар М., де Виссер М., Шманд Б.А., де Хаан Р.Дж. (июнь 2016 г.). «Когнитивный профиль БАС: систематический обзор и обновление метаанализа». Журнал неврологии, нейрохирургии и психиатрии . 87 (6): 611–619. дои : 10.1136/jnnp-2015-310734 . ПМИД 26283685 . S2CID 22082109 .
- ^ Перейти обратно: а б с д Аль-Чалаби А., Хардиман О (ноябрь 2013 г.). «Эпидемиология БАС: заговор генов, окружающей среды и времени». Обзоры природы. Неврология . 9 (11): 617–628. дои : 10.1038/nrneurol.2013.203 . ПМИД 24126629 . S2CID 25040863 .
- ^ Перейти обратно: а б «Боковой амиотрофический склероз (БАС) – симптомы и причины» . Клиника Мэйо . Архивировано из оригинала 6 апреля 2022 года . Проверено 6 апреля 2022 г.
- ^ Перейти обратно: а б с «Кто болеет БАС?» . Ассоциация БАС . Архивировано из оригинала 6 апреля 2022 года . Проверено 6 апреля 2022 г.
- ^ Хе Дж., Мангельсдорф М., Фан Д., Бартлетт П., Браун М.А. (декабрь 2015 г.). «Генетические исследования бокового амиотрофического склероза: от картирования общегеномных ассоциаций к секвенированию генома» (PDF) . Нейробиолог . 21 (6): 599–615. дои : 10.1177/1073858414555404 . ПМИД 25378359 . S2CID 3437565 . Архивировано (PDF) из оригинала 7 мая 2020 года . Проверено 20 декабря 2019 г.
- ^ Макнил А., Амадор М.Д., Беккер Х., Кларк А., Крук А., Каммингс С. и др. (июнь 2022 г.). «Прогностическое генетическое тестирование заболеваний двигательных нейронов: время для рекомендаций?» . Европейский журнал генетики человека . 30 (6): 635–636. дои : 10.1038/s41431-022-01093-y . ПМЦ 9177585 . ПМИД 35379930 .
- ^ Де Оливейра Х.М., Сома А., Бейкер М.Р., Тернер М.Р., Талбот К., Уильямс Т.Л. (август 2023 г.). «Обзор текущей практики генетического тестирования при боковом амиотрофическом склерозе в Великобритании и Ирландии: последствия для будущего планирования» . Боковой амиотрофический склероз и лобно-височная дегенерация . 24 (5–6): 405–413. дои : 10.1080/21678421.2022.2150556 . ПМИД 36458618 . S2CID 254150195 .
- ^ Мюллер К., О К.В., Нордин А., Панти С., Ким Ш., Нордин Ф. и др. (февраль 2022 г.). «Мутации de novo в SOD1 являются причиной БАС» . Журнал неврологии, нейрохирургии и психиатрии . 93 (2): 201–206. дои : 10.1136/jnnp-2021-327520 . ПМЦ 8784989 . ПМИД 34518333 .
- ^ Макнил А., Амадор М.Д., Беккер Х., Кларк А., Крук А., Каммингс С. и др. (Международный альянс ассоциаций БАС/БДН) (июнь 2022 г.). «Прогностическое генетическое тестирование заболеваний двигательных нейронов: время для рекомендаций?» . Европейский журнал генетики человека . 30 (6): 635–636. дои : 10.1038/s41431-022-01093-y . ПМЦ 9177585 . ПМИД 35379930 .
- ^ Салмон К., Кирнан М.К., Ким Ш., Андерсен П.М., Чио А., ван ден Берг Л.Х. и др. (май 2022 г.). «Важность раннего генетического тестирования у всех больных боковым амиотрофическим склерозом» . Мозг . 145 (4): 1207–1210. дои : 10.1093/brain/awab472 . ПМК 9129091 . ПМИД 35020823 . Архивировано из оригинала 27 мая 2023 года . Проверено 27 мая 2023 г.
- ^ Перейти обратно: а б с Ван, доктор медицинских наук, Литтл Дж., Гомес Дж., Кэшман Н.Р., Кревски Д. (июль 2017 г.). «Идентификация факторов риска, связанных с возникновением и прогрессированием бокового амиотрофического склероза, с использованием систематического обзора и метаанализа» . Нейротоксикология . 61 : 101–130. Бибкод : 2017NeuTx..61..101W . дои : 10.1016/j.neuro.2016.06.015 . ПМИД 27377857 . S2CID 33604904 . Архивировано из оригинала 17 ноября 2022 года . Проверено 27 мая 2023 г.
- ^ Перейти обратно: а б Гроссман А.Б., Левин Б.Е., Брэдли В.Г. (март 2006 г.). «Преморбидные особенности личности больных БАС». Боковой амиотрофический склероз . 7 (1): 27–31. дои : 10.1080/14660820510012004 . ПМИД 16546756 . S2CID 20998807 .
- ^ Перейти обратно: а б с Паркин Куллманн Дж. А., Хейс С., Памфлетт Р. (октябрь 2018 г.). «Являются ли люди с боковым амиотрофическим склерозом (БАС) особенно приятными? Международное онлайн-исследование «случай-контроль» Большой Пятерки личностных факторов» . Мозг и поведение . 8 (10): e01119. дои : 10.1002/brb3.1119 . ПМК 6192405 . ПМИД 30239176 .
- ^ Мель Т., Джордан Б., Зирц С. (январь 2017 г.). « Пациенты с боковым амиотрофическим склерозом (АЛС) обычно приятные люди» — Как врачи, страдающие БАС, видят личностные особенности своих пациентов» . Мозг и поведение . 7 (1): e00599. дои : 10.1002/brb3.599 . ПМК 5256182 . ПМИД 28127517 .
- ^ Робберехт В., Филипс Т. (апрель 2013 г.). «Изменяющаяся сцена бокового амиотрофического склероза». Обзоры природы. Нейронаука . 14 (4): 248–264. дои : 10.1038/nrn3430 . ПМИД 23463272 . S2CID 208941 .
- ^ Перейти обратно: а б с д и ж г час Браун Р.Х., Аль-Чалаби А. (июль 2017 г.). «Боковой амиотрофический склероз» . Медицинский журнал Новой Англии . 377 (2): 162–172. дои : 10.1056/NEJMra1603471 . ПМИД 28700839 . S2CID 205117619 . Архивировано из оригинала 25 февраля 2021 года . Проверено 20 декабря 2019 г.
- ^ Окамото К., Мизуно Ю., Фудзита Ю. (апрель 2008 г.). «Тельца Бунина при боковом амиотрофическом склерозе». Невропатология . 28 (2): 109–115. дои : 10.1111/j.1440-1789.2007.00873.x . ПМИД 18069968 . S2CID 34398467 .
- ^ Уайт Дж.А., Банерджи Р., Гунавардена С. (май 2016 г.). «Аксональный транспорт и нейродегенерация: как морские препараты можно использовать для разработки терапии» . Морские наркотики . 14 (5): 102. дои : 10.3390/md14050102 . ПМЦ 4882576 . ПМИД 27213408 .
- ^ Нг Ки Квонг К.К., Мехта А.Р., Недергаард М., Чандран С. (август 2020 г.). «Определение новых функций спинномозговой жидкости в патофизиологии БАС» . Acta Neuropathologica Communications . 8 (1): 140. дои : 10.1186/s40478-020-01018-0 . ПМЦ 7439665 . ПМИД 32819425 .
- ^ Ван Дамм П., Робберехт В., Ван Ден Бош Л. (май 2017 г.). «Моделирование бокового амиотрофического склероза: прогресс и возможности» . Модели и механизмы заболеваний . 10 (5): 537–549. дои : 10.1242/dmm.029058 . ПМЦ 5451175 . ПМИД 28468939 .
- ^ Алсултан А.А., Уоллер Р., Хит П.Р., Кирби Дж. (2016). «Генетика бокового амиотрофического склероза: современные представления» . Дегенеративные неврологические и нервно-мышечные заболевания . 6 : 49–64. дои : 10.2147/DNND.S84956 . ПМК 6053097 . ПМИД 30050368 .
- ^ Шан Ю, Хуан Э.Дж. (сентябрь 2016 г.). «Механизмы мутаций FUS при семейном боковом амиотрофическом склерозе» . Исследования мозга . 1647 : 65–78. дои : 10.1016/j.brainres.2016.03.036 . ПМК 5003642 . ПМИД 27033831 .
- ^ Нгуен Х.П., Ван Брокховен С., ван дер Зи Дж. (июнь 2018 г.). «Гены БАС в эпоху геномики и их значение для ЛВД» . Тенденции в генетике . 34 (6): 404–423. дои : 10.1016/j.tig.2018.03.001 . hdl : 10067/1514730151162165141 . ПМИД 29605155 .
- ^ Гитлер А.Д., Цуйджи Х. (сентябрь 2016 г.). «Произошло пробуждение: новые механизмы мутаций C9orf72 при ЛВД/БАС» . Исследования мозга . 1647 : 19–29. дои : 10.1016/j.brainres.2016.04.004 . ПМК 5003651 . ПМИД 27059391 .
- ^ Мехта А.Р., Грегори Дж.М., Дандо О., Картер Р.Н., Берр К., Нанда Дж. и др. (февраль 2021 г.). «Митохондриальный биоэнергетический дефицит в мотонейронах бокового амиотрофического склероза C9orf72 вызывает дисфункциональный аксональный гомеостаз» . Акта Нейропатологика . 141 (2): 257–279. дои : 10.1007/s00401-020-02252-5 . ПМЦ 7847443 . ПМИД 33398403 .
- ^ Вербер Н.С., Шепард С.Р., Сассани М., Макдонаф Х.Э., Мур С.А., Аликс Дж.Дж. и др. (2019). «Биомаркеры заболеваний двигательных нейронов: современный обзор» . Границы в неврологии . 10 : 291. дои : 10.3389/fneur.2019.00291 . ПМК 6456669 . ПМИД 31001186 .
- ^ Джойс, Северная Каролина, Картер Г.Т. (май 2013 г.). «Электродиагностика у лиц с боковым амиотрофическим склерозом» . ПМиР . 5 (5 доп.): S89–S95. дои : 10.1016/j.pmrj.2013.03.020 . ПМК 4590769 . ПМИД 23523708 .
- ^ Силани В., Мессина С., Полетти Б., Морелли С., Доретти А., Тикоцци Н., Мадерна Л. (март 2011 г.). «Диагноз боковой амиотрофический склероз в 2010 году». Archives Italiennes de Biologie . 149 (1): 5–27. дои : 10.4449/aib.v149i1.1260 . ПМИД 21412713 .
- ^ Стиклер Д.Э. Лоренцо Н. (ред.). «Миастенический синдром Ламберта-Итона (LEMS)» . Разное.medscape.com. Архивировано из оригинала 14 мая 2013 года . Проверено 18 апреля 2013 г.
- ^ «Миастенический синдром Ламберта-Итона: О» . LEMS.com . BioMarin Pharmaceutical Inc. Архивировано из оригинала 20 января 2013 года . Проверено 18 апреля 2013 г.
- ^ Миллс КР (ноябрь 2010 г.). «Характеристика фасцикуляций при боковом амиотрофическом склерозе и синдроме доброкачественных фасцикуляций» . Мозг . 133 (11): 3458–3469. дои : 10.1093/brain/awq290 . ПМИД 20959307 .
- ^ Перейти обратно: а б Радунович А., Аннан Д., Рафик МК, Брассингтон Р., Мустфа Н. (октябрь 2017 г.). «Механическая вентиляция при боковом амиотрофическом склерозе/заболевании двигательных нейронов» . Кокрановская база данных систематических обзоров . 10 (10): CD004427. дои : 10.1002/14651858.CD004427.pub4 . ПМЦ 6485636 . ПМИД 28982219 .
- ^ Перейти обратно: а б с д Ахмед Р.М., Ньюкомб Р.Э., Пайпер А.Дж., Льюис С.Дж., Йи Б.Дж., Кирнан М.К., Грунштейн Р.Р. (апрель 2016 г.). «Нарушения сна и дыхательная функция при боковом амиотрофическом склерозе». Обзоры медицины сна . 26 : 33–42. дои : 10.1016/j.smrv.2015.05.007 . ПМИД 26166297 .
- ^ Перейти обратно: а б Эйзен А., Кригер С. (ноябрь 2013 г.). «Этические соображения при лечении бокового амиотрофического склероза». Прогресс нейробиологии . 110 : 45–53. doi : 10.1016/j.pneurobio.2013.05.001 . ПМИД 23735671 . S2CID 26282198 .
- ^ Перейти обратно: а б Льюис М., Рушанан С. (2007). «Роль физиотерапии и трудотерапии в лечении бокового амиотрофического склероза». Нейрореабилитация . 22 (6): 451–461. дои : 10.3233/NRE-2007-22608 . ПМИД 18198431 .
- ^ Перейти обратно: а б с д и «Боковой амиотрофический склероз (АЛС)» . Американская ассоциация речи, языка и слуха, Роквилл, Мэриленд. Архивировано из оригинала 2 августа 2012 года . Проверено 30 ноября 2016 г. .
- ^ Перейти обратно: а б с Оррелл Р.В. (2010). «Болезнь двигательных нейронов: систематические обзоры лечения БАС и СМА» . Британский медицинский бюллетень . 93 : 145–159. дои : 10.1093/bmb/ldp049 . ПМИД 20015852 .
- ^ Перейти обратно: а б с Арбесман М., Шеард К. (2014). «Систематический обзор эффективности вмешательств, связанных с эрготерапией, у людей с боковым амиотрофическим склерозом» . Американский журнал профессиональной терапии . 68 (1): 20–26. дои : 10.5014/ajot.2014.008649 . ПМИД 24367951 .
- ^ Перейти обратно: а б Сулистё А., Абрахао А., Фрейтас М.Э., Ритсма Б., Зинман Л. (август 2023 г.). «Энтеральное зондовое питание при боковом амиотрофическом склерозе/заболевании двигательных нейронов». Кокрановская база данных систематических обзоров . 2023 (8): CD004030. дои : 10.1002/14651858.CD004030.pub4 . PMC 10413437. PMID 37579081 .
- ^ Перейти обратно: а б с Андерсен П.М., Абрахамс С., Борасио Г.Д., де Карвальо М., Чио А., Ван Дамм П. и др. (март 2012 г.). «Руководство EFNS по клиническому лечению бокового амиотрофического склероза (MALS) — пересмотренный отчет целевой группы EFNS» . Европейский журнал неврологии . 19 (3): 360–375. дои : 10.1111/j.1468-1331.2011.03501.x . ПМИД 21914052 . S2CID 5746940 .
- ^ Перейти обратно: а б с д Миллер Р.Г., Митчелл Дж.Д., Мур Д.Х. (март 2012 г.). «Рилузол при боковом амиотрофическом склерозе (БАС)/заболевании двигательных нейронов (БДН)» . Кокрановская база данных систематических обзоров . 2012 (3): CD001447. дои : 10.1002/14651858.CD001447.pub3 . ПМК 7055506 . ПМИД 22419278 .
- ^ Карлези С., Паскуали Л., Пьяцца С., Ло Герфо А., Кальдараццо Иенко Э., Алесси Р. и др. (март 2011 г.). «Стратегии клинического подхода к нейродегенерации при боковом амиотрофическом склерозе». Archives Italiennes de Biologie . 149 (1): 151–167. дои : 10.4449/aib.v149i1.1267 . ПМИД 21412722 .
- ^ Абэ, Кодзи; и др. (июль 2017 г.). «Безопасность и эффективность эдаравона у четко определенных пациентов с боковым амиотрофическим склерозом: рандомизированное двойное слепое плацебо-контролируемое исследование» . «Ланцет». Неврология . 16 (7): 505–512. дои : 10.1016/S1474-4422(17)30115-1 . ПМИД 28522181 .
- ^ Перейти обратно: а б с д и Дорст Дж., Людольф AC, Хьюберс А. (2018). «Модифицирующее заболевание и симптоматическое лечение бокового амиотрофического склероза» . Терапевтические достижения в области неврологических расстройств . 11 : 1756285617734734. дои : 10.1177/1756285617734734 . ПМК 5784546 . ПМИД 29399045 .
- ^ Такей К., Ватанабэ К., Юки С., Акимото М., Саката Т., Палумбо Дж. (октябрь 2017 г.). «Эдаравон и его клиническое развитие при боковом амиотрофическом склерозе» . Боковой амиотрофический склероз и лобно-височная дегенерация . 18 (суп1): 5–10. дои : 10.1080/21678421.2017.1353101 . ПМИД 28872907 .
- ^ Центр оценки и исследований лекарств (16 июня 2022 г.). «FDA одобрило пероральную форму для лечения бокового амиотрофического склероза (БАС) у взрослых» . Управление по контролю за продуктами и лекарствами США. Архивировано из оригинала 12 мая 2022 года . Проверено 25 апреля 2023 г.
- ^ «AMX0035 (РЕЛИВРИО)» . Ассоциация БАС . Архивировано из оригинала 25 апреля 2023 года . Проверено 25 апреля 2023 г.
- ^ «Лекарство от АЛС будет снято с рынка США после того, как исследование показало, что оно не приносит пользы пациентам» . АП Новости . 4 апреля 2024 г. Проверено 4 апреля 2024 г.
- ^ Amylyx Pharmaceuticals Inc. (4 января 2023 г.). Фаза III, рандомизированное двойное слепое плацебо-контролируемое многоцентровое исследование для оценки безопасности и эффективности AMX0035 по сравнению с плацебо для 48-недельного лечения взрослых пациентов с боковым амиотрофическим склерозом (БАС) (отчет). www.clinicaltrials.gov.
- ^ Перейти обратно: а б «FDA одобрило лечение бокового амиотрофического склероза, связанного с мутацией гена SOD1» (Пресс-релиз). США Управление по контролю за продуктами и лекарствами (FDA). 25 апреля 2023 года. Архивировано из оригинала 25 апреля 2023 года . Проверено 25 апреля 2023 г.
- ^ Миллер Т.М., Кудкович М.Е., Джендж А., Шоу П.Дж., Собу Г., Бучелли Р.К. и др. (сентябрь 2022 г.). «Испытание антисмыслового олигонуклеотида Тоферсена при SOD1 БАС» . Медицинский журнал Новой Англии . 387 (12): 1099–1110. дои : 10.1056/NEJMoa2204705 . ПМИД 36129998 . S2CID 252438252 .
- ^ Лу Ч., Макдональд-Уоллис С., Грей Е., Пирс Н., Петцольд А., Норгрен Н. и др. (июнь 2015 г.). «Легкая цепь нейрофиламента: прогностический биомаркер при боковом амиотрофическом склерозе» . Неврология . 84 (22): 2247–2257. дои : 10.1212/WNL.0000000000001642 . ПМЦ 4456658 . ПМИД 25934855 .
- ^ Перейти обратно: а б Паганони С., Карам С., Джойс Н., Бедлак Р., Картер Г.Т. (2015). «Комплексная реабилитационная помощь по всему спектру бокового амиотрофического склероза» . Нейрореабилитация . 37 (1): 53–68. дои : 10.3233/NRE-151240 . ПМЦ 5223769 . ПМИД 26409693 .
- ^ Пинту С., Карвальо М. (2014). «Вдохнуть новую жизнь в достижения в лечении дыхательной недостаточности у пациентов с боковым амиотрофическим склерозом». Управление нейродегенеративными заболеваниями . 4 (1): 83–102. дои : 10.2217/nmt.13.74 . ПМИД 24640982 .
- ^ Маджмудар С., Ву Дж., Паганони С. (июль 2014 г.). «Реабилитация при боковом амиотрофическом склерозе: почему это важно» . Мышцы и нервы . 50 (1): 4–13. дои : 10.1002/mus.24202 . ПМК 4433000 . ПМИД 24510737 .
- ^ Макферсон CE, Bassile CC (июль 2016 г.). «Методы легочной физиотерапии для повышения выживаемости при боковом амиотрофическом склерозе: систематический обзор». Журнал неврологической физиотерапии . 40 (3): 165–175. дои : 10.1097/NPT.0000000000000136 . ПМИД 27164308 . S2CID 7279853 .
- ^ Спатаро Р., Ло Ре М., Пикколи Т., Пикколи Ф., Ла Белла В. (сентябрь 2010 г.). «Причины и место смерти итальянских больных боковым амиотрофическим склерозом» . Acta Neurologica Scandinavica . 122 (3): 217–223. дои : 10.1111/j.1600-0404.2009.01290.x . ПМИД 20078446 . S2CID 26479278 .
- ^ Чековей Х., Лундин Дж.И., Келада С.Н. (2011). «Глава 22: Нейродегенеративные заболевания». Ротман Н., Эно П., Шульте П., Смит М., Боффетта П., Перера Ф. (ред.). Молекулярная эпидемиология: принципы и практика . Международное агентство по исследованию рака. стр. 408–409. ISBN 978-9283221630 .
- ^ Чио А., Логроскино Дж., Трейнор Б.Дж., Коллинз Дж., Симеоне Дж.К., Гольдштейн Л.А., Уайт Л.А. (2013). «Глобальная эпидемиология бокового амиотрофического склероза: систематический обзор опубликованной литературы» . Нейроэпидемиология . 41 (2): 118–130. дои : 10.1159/000351153 . ПМК 4049265 . ПМИД 23860588 .
- ^ Хардиман О., Аль-Чалаби А., Брейн С., Беги Э., ван ден Берг Л.Х., Чио А. и др. (июль 2017 г.). «Изменяющаяся картина бокового амиотрофического склероза: уроки европейских регистров» . Журнал неврологии, нейрохирургии и психиатрии . 88 (7): 557–563. дои : 10.1136/jnnp-2016-314495 . hdl : 2318/1633611 . ПМИД 28285264 . S2CID 52871105 . Архивировано из оригинала 21 декабря 2019 года . Проверено 22 августа 2018 г.
- ^ Перейти обратно: а б с д и Мехта П., Кэй В., Рэймонд Дж., Панджани Р., Ларсон Т., Коэн Дж. и др. (ноябрь 2018 г.). «Распространенность бокового амиотрофического склероза – США, 2015 г.» . ММВР. Еженедельный отчет о заболеваемости и смертности . 67 (46): 1285–1289. дои : 10.15585/mmwr.mm6746a1 . ПМЦ 5858037 . ПМИД 30462626 .
- ^ Перейти обратно: а б Артур К.К., Кальво А., Прайс Т.Р., Гейгер Дж.Т., Чио А., Трейнор Б.Дж. (август 2016 г.). «Прогнозируемый рост заболеваемости боковым амиотрофическим склерозом с 2015 по 2040 год» . Природные коммуникации . 7 (12408): 12408. Бибкод : 2016NatCo...712408A . дои : 10.1038/ncomms12408 . ПМЦ 4987527 . ПМИД 27510634 .
- ^ Перейти обратно: а б Луна Дж., Логроскино Дж., Куратье П., Марин Б. (май 2017 г.). «Актуальные проблемы эпидемиологии БАС: различия в распространенности БАС между группами населения и физическая активность как фактор риска». Ревю Неврологии . 173 (5): 244–253. дои : 10.1016/j.neurol.2017.03.035 . ПМИД 28477849 .
- ^ Цзоу З.И., Чжоу З.Р., Че Ч., Лю С.И., Хэ Р.Л., Хуан Х.П. (июль 2017 г.). «Генетическая эпидемиология бокового амиотрофического склероза: систематический обзор и метаанализ». Журнал неврологии, нейрохирургии и психиатрии . 88 (7): 540–549. дои : 10.1136/jnnp-2016-315018 . ПМИД 28057713 . S2CID 41974606 .
- ^ Перейти обратно: а б Тейве Х.А., Лима ПМ, Джерминиани ФМ, Муньоз РП (май 2016 г.). «Что в имени? Проблемы, факты и споры относительно неврологических эпонимов» . Arquivos de Neuro-Psiquiatria . 74 (5): 423–425. дои : 10.1590/0004-282X20160040 . ПМИД 27191240 .
- ^ Виссер Дж., де Йонг Дж. М., де Виссер М. (февраль 2008 г.). «История прогрессирующей мышечной атрофии: синдром или болезнь?». Неврология . 70 (9): 723–727. дои : 10.1212/01.wnl.0000302187.20239.93 . ПМИД 18299524 . S2CID 22629725 .
- ^ Виджесекера Л.С., Мазерс С., Талман П., Галтри С., Паркинсон М.Х., Ганесалингам Дж. и др. (март 2009 г.). «Естественное течение и клинические особенности вариантов БАС с цеповой рукой и цеповой ногой» . Неврология . 72 (12): 1087–1094. дои : 10.1212/01.wnl.0000345041.83406.a2 . ПМК 2821838 . ПМИД 19307543 .
- ^ Уилборн Эй Джей (октябрь 1998 г.). «Клиническая нейрофизиология в диагностике бокового амиотрофического склероза: критерии Ламберта и Эль-Эскориала». Журнал неврологических наук . 160 (Приложение 1): С25–С29. дои : 10.1016/s0022-510x(98)00194-4 . ПМИД 9851644 . S2CID 32884687 .
- ^ Брукс БР (июль 1994 г.). «Критерии Всемирной федерации неврологии Эль-Эскориала для диагностики бокового амиотрофического склероза. Подкомитет по заболеваниям двигательных нейронов / боковому амиотрофическому склерозу Исследовательской группы по нейромышечным заболеваниям Всемирной федерации неврологии и участники семинара Эль-Эскориал «Клинические пределы бокового амиотрофического склероза». ". Журнал неврологических наук . 124 (Приложение): 96–107. дои : 10.1016/0022-510x(94)90191-0 . ПМИД 7807156 . S2CID 32678612 .
- ^ Брукс Б.Р., Миллер Р.Г., Сваш М., Мунсат Т.Л. (декабрь 2000 г.). «Возвращение к Эль-Эскориалу: пересмотренные критерии диагностики бокового амиотрофического склероза». Боковой амиотрофический склероз и другие заболевания двигательных нейронов . 1 (5): 293–299. дои : 10.1080/146608200300079536 . PMID 11464847 . S2CID 22725949 .
- ^ де Карвальо М., Денглер Р., Эйзен А., Англия Дж.Д., Кадзи Р., Кимура Дж. и др. (март 2008 г.). «Электродиагностические критерии диагностики БАС». Клиническая нейрофизиология . 119 (3): 497–503. дои : 10.1016/j.clinph.2007.09.143 . ПМИД 18164242 . S2CID 14851649 .
- ^ Шефнер Дж.М., Аль-Чалаби А., Бейкер М.Р., Кюи Л.И., де Карвалью М., Эйзен А. и др. (август 2020 г.). «Предложение о новых диагностических критериях БАС» . Клиническая нейрофизиология . 131 (8): 1975–1978. doi : 10.1016/j.clinph.2020.04.005 . hdl : 10451/48432 . ПМИД 32387049 . S2CID 215823371 .
- ^ Перейти обратно: а б «Что такое БАС?» . Ассоциация БАС . Архивировано из оригинала 21 декабря 2018 года . Проверено 23 декабря 2018 г.
- ^ «АЛС: боковой амиотрофический склероз» . Ассоциация мышечной дистрофии . 18 декабря 2015 г. Архивировано из оригинала 6 августа 2018 г. Проверено 23 декабря 2018 г.
- ^ Гетц К.Г. (март 2000 г.). «Боковой амиотрофический склероз: ранние вклады Жана-Мартена Шарко». Мышцы и нервы . 23 (3): 336–343. doi : 10.1002/(SICI)1097-4598(200003)23:3<336::AID-MUS4>3.0.CO;2-L . ПМИД 10679709 . S2CID 5917354 .
- ^ Гордон П.Х. (2006). «Глава 1: История БАС». В Мицумото Х., Пшедборски С., Гордон П.Х. (ред.). Боковой амиотрофический склероз . ЦРК Пресс. п. 9. ISBN 978-0824729240 .
- ^ Гордон П.Х. (октябрь 2013 г.). «Боковой амиотрофический склероз: обновленная информация о клинических особенностях, патофизиологии, ведении и терапевтических исследованиях 2013 года» . Старение и болезни . 4 (5): 295–310. дои : 10.14336/AD.2013.0400295 . ПМЦ 3794725 . ПМИД 24124634 .
- ^ Перейти обратно: а б Хюинь В., Саймон Н.Г., Гросскройц Дж., Тернер М.Р., Вучич С., Кирнан М.К. (июль 2016 г.). «Оценка верхнего мотонейрона при боковом амиотрофическом склерозе» . Клиническая нейрофизиология . 127 (7): 2643–2660. дои : 10.1016/j.clinph.2016.04.025 . ПМИД 27291884 . S2CID 3757685 .
- ^ Александр Э (20 августа 2014 г.). «Джордж Буш представляет, возможно, лучший вызов ведерку со льдом для БАС» . Независимый . Архивировано из оригинала 21 августа 2014 года . Проверено 20 августа 2014 г.
- ^ Уикс П. (декабрь 2014 г.). «Испытание ведра со льдом при БАС – может ли брызги воды оживить поле?» . Боковой амиотрофический склероз и лобно-височная дегенерация . 15 (7–8): 479–480. дои : 10.3109/21678421.2014.984725 . ПМИД 25431828 . S2CID 207581186 .
Внешние ссылки
[ редактировать ]![]() СМИ, связанные с боковым амиотрофическим склерозом, на Викискладе?
СМИ, связанные с боковым амиотрофическим склерозом, на Викискладе?