Клиническое исследование

Клинические испытания — это проспективные биомедицинские или поведенческие исследования на людях, призванные ответить на конкретные вопросы о биомедицинских или поведенческих вмешательствах, включая новые методы лечения (такие как новые вакцины , лекарства , диетические предпочтения , пищевые добавки и медицинские устройства ) и известные вмешательства, которые требуют дальнейшего изучение и сравнение. Клинические испытания собирают данные о дозировке, безопасности и эффективности. [1] [2] Они проводятся только после получения одобрения органа здравоохранения/комитета по этике в стране, где запрашивается одобрение терапии. Эти органы несут ответственность за проверку соотношения риска и пользы в исследовании — их одобрение не означает, что терапия «безопасна» или эффективна, а означает лишь то, что исследование может быть проведено.
В зависимости от типа продукта и стадии разработки исследователи сначала привлекают добровольцев или пациентов к небольшим пилотным исследованиям , а затем проводят сравнительные исследования все большего масштаба. Клинические испытания могут различаться по размеру и стоимости, и в них может участвовать один исследовательский центр или несколько центров в одной стране или в нескольких странах. Дизайн клинического исследования направлен на обеспечение научной достоверности и воспроизводимости результатов.
Costs for clinical trials can range into the billions of dollars per approved drug,[3] and the complete trial process to approval may require 7–15 years.[4][5] The sponsor may be a governmental organization or a pharmaceutical, biotechnology or medical-device company. Certain functions necessary to the trial, such as monitoring and lab work, may be managed by an outsourced partner, such as a contract research organization or a central laboratory. Only 10 percent of all drugs started in human clinical trials become approved drugs.[6]
Overview[edit]
Trials of drugs[edit]
Some clinical trials involve healthy subjects with no pre-existing medical conditions. Other clinical trials pertain to people with specific health conditions who are willing to try an experimental treatment. Pilot experiments are conducted to gain insights for design of the clinical trial to follow.
There are two goals to testing medical treatments: to learn whether they work well enough, called "efficacy", or "effectiveness"; and to learn whether they are safe enough, called "safety".[1] Neither is an absolute criterion; both safety and efficacy are evaluated relative to how the treatment is intended to be used, what other treatments are available, and the severity of the disease or condition. The benefits must outweigh the risks.[7][8]: 8 For example, many drugs to treat cancer have severe side effects that would not be acceptable for an over-the-counter pain medication, yet the cancer drugs have been approved since they are used under a physician's care and are used for a life-threatening condition.[9]
In the US the elderly constitute 14% of the population, while they consume over one-third of drugs.[10] People over 55 (or a similar cutoff age) are often excluded from trials because their greater health issues and drug use complicate data interpretation, and because they have different physiological capacity than younger people. Children and people with unrelated medical conditions are also frequently excluded.[11] Pregnant women are often excluded due to potential risks to the fetus.
The sponsor designs the trial in coordination with a panel of expert clinical investigators, including what alternative or existing treatments to compare to the new drug and what type(s) of patients might benefit. If the sponsor cannot obtain enough test subjects at one location investigators at other locations are recruited to join the study.
During the trial, investigators recruit subjects with the predetermined characteristics, administer the treatment(s) and collect data on the subjects' health for a defined time period. Data include measurements such as vital signs, concentration of the study drug in the blood or tissues, changes to symptoms, and whether improvement or worsening of the condition targeted by the study drug occurs. The researchers send the data to the trial sponsor, who then analyzes the pooled data using statistical tests.
Examples of clinical trial goals include assessing the safety and relative effectiveness of a medication or device:
- On a specific kind of patient
- At varying dosages
- For a new indication
- Evaluation for improved efficacy in treating a condition as compared to the standard therapy for that condition
- Evaluation of the study drug or device relative to two or more already approved/common interventions for that condition
While most clinical trials test one alternative to the novel intervention, some expand to three or four and may include a placebo.
Except for small, single-location trials, the design and objectives are specified in a document called a clinical trial protocol. The protocol is the trial's "operating manual" and ensures all researchers perform the trial in the same way on similar subjects and that the data is comparable across all subjects.
As a trial is designed to test hypotheses and rigorously monitor and assess outcomes, it can be seen as an application of the scientific method, specifically the experimental step.
The most common clinical trials evaluate new pharmaceutical products, medical devices, biologics, diagnostic assays, psychological therapies, or other interventions.[12] Clinical trials may be required before a national regulatory authority[13] approves marketing of the innovation.
Trials of devices[edit]
Similarly to drugs, manufacturers of medical devices in the United States are required to conduct clinical trials for premarket approval.[14] Device trials may compare a new device to an established therapy, or may compare similar devices to each other. An example of the former in the field of vascular surgery is the Open versus Endovascular Repair (OVER trial) for the treatment of abdominal aortic aneurysm, which compared the older open aortic repair technique to the newer endovascular aneurysm repair device.[15] An example of the latter are clinical trials on mechanical devices used in the management of adult female urinary incontinence.[16]
Trials of procedures[edit]
Similarly to drugs, medical or surgical procedures may be subjected to clinical trials,[17] such as comparing different surgical approaches in treatment of fibroids for subfertility.[18] However, when clinical trials are unethical or logistically impossible in the surgical setting, case-controlled studies will be replaced.[19]
Patient and public involvement[edit]
Besides being participants in a clinical trial, members of the public can be actively collaborate with researchers in designing and conducting clinical research. This is known as patient and public involvement (PPI). Public involvement involves a working partnership between patients, caregivers, people with lived experience, and researchers to shape and influence what is researcher and how.[20] PPI can improve the quality of research and make it more relevant and accessible. People with current or past experience of illness can provide a different perspective than professionals and compliment their knowledge. Through their personal knowledge they can identify research topics that are relevant and important to those living with an illness or using a service. They can also help to make the research more grounded in the needs of the specific communities they are part of. Public contributors can also ensure that the research is presented in plain language that is clear to the wider society and the specific groups it is most relevant for.[21]
History[edit]
Development[edit]
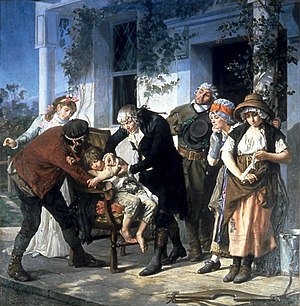
Although early medical experimentation was performed often, the use of a control group to provide an accurate comparison for the demonstration of the intervention's efficacy was generally lacking. For instance, Lady Mary Wortley Montagu, who campaigned for the introduction of inoculation (then called variolation) to prevent smallpox, arranged for seven prisoners who had been sentenced to death to undergo variolation in exchange for their life. Although they survived and did not contract smallpox, there was no control group to assess whether this result was due to the inoculation or some other factor. Similar experiments performed by Edward Jenner over his smallpox vaccine were equally conceptually flawed.[22]
The first proper clinical trial was conducted by the Scottish physician James Lind.[23] The disease scurvy, now known to be caused by a Vitamin C deficiency, would often have terrible effects on the welfare of the crew of long-distance ocean voyages. In 1740, the catastrophic result of Anson's circumnavigation attracted much attention in Europe; out of 1900 men, 1400 had died, most of them allegedly from having contracted scurvy.[24] John Woodall, an English military surgeon of the British East India Company, had recommended the consumption of citrus fruit (it has an antiscorbutic effect) from the 17th century, but their use did not become widespread.[25]
Lind conducted the first systematic clinical trial in 1747.[26] He included a dietary supplement of an acidic quality in the experiment after two months at sea, when the ship was already afflicted with scurvy. He divided twelve scorbutic sailors into six groups of two. They all received the same diet but, in addition, group one was given a quart of cider daily, group two twenty-five drops of elixir of vitriol (sulfuric acid), group three six spoonfuls of vinegar, group four half a pint of seawater, group five received two oranges and one lemon, and the last group a spicy paste plus a drink of barley water. The treatment of group five stopped after six days when they ran out of fruit, but by then one sailor was fit for duty while the other had almost recovered. Apart from that, only group one also showed some effect of its treatment.[27] Each year, May 20 is celebrated as Clinical Trials Day in honor of Lind's research.[28]
After 1750 the discipline began to take its modern shape.[29][30] The English doctor John Haygarth demonstrated the importance of a control group for the correct identification of the placebo effect in his celebrated study of the ineffective remedy called Perkin's tractors. Further work in that direction was carried out by the eminent physician Sir William Gull, 1st Baronet in the 1860s.[22]
Frederick Akbar Mahomed (d. 1884), who worked at Guy's Hospital in London, made substantial contributions to the process of clinical trials, where "he separated chronic nephritis with secondary hypertension from what we now term essential hypertension. He also founded the Collective Investigation Record for the British Medical Association; this organization collected data from physicians practicing outside the hospital setting and was the precursor of modern collaborative clinical trials."[31]
Modern trials[edit]

Sir Ronald A. Fisher, while working for the Rothamsted experimental station in the field of agriculture, developed his Principles of experimental design in the 1920s as an accurate methodology for the proper design of experiments. Among his major ideas, was the importance of randomization—the random assignment of individuals to different groups for the experiment;[32] replication—to reduce uncertainty, measurements should be repeated and experiments replicated to identify sources of variation;[33] blocking—to arrange experimental units into groups of units that are similar to each other, and thus reducing irrelevant sources of variation; use of factorial experiments—efficient at evaluating the effects and possible interactions of several independent factors.[22]
The British Medical Research Council officially recognized the importance of clinical trials from the 1930s. The council established the Therapeutic Trials Committee to advise and assist in the arrangement of properly controlled clinical trials on new products that seem likely on experimental grounds to have value in the treatment of disease.[22]
The first randomised curative trial was carried out at the MRC Tuberculosis Research Unit by Sir Geoffrey Marshall (1887–1982). The trial, carried out between 1946 and 1947, aimed to test the efficacy of the chemical streptomycin for curing pulmonary tuberculosis. The trial was both double-blind and placebo-controlled.[34]
The methodology of clinical trials was further developed by Sir Austin Bradford Hill, who had been involved in the streptomycin trials. From the 1920s, Hill applied statistics to medicine, attending the lectures of renowned mathematician Karl Pearson, among others. He became famous for a landmark study carried out in collaboration with Richard Doll on the correlation between smoking and lung cancer. They carried out a case-control study in 1950, which compared lung cancer patients with matched control and also began a sustained long-term prospective study into the broader issue of smoking and health, which involved studying the smoking habits and health of more than 30,000 doctors over a period of several years. His certificate for election to the Royal Society called him "... the leader in the development in medicine of the precise experimental methods now used nationally and internationally in the evaluation of new therapeutic and prophylactic agents."
International clinical trials day is celebrated on 20 May.[35]
The acronyms used in the titling of clinical trials is often contrived, and has been the subject of derision.[36]
Types[edit]
Clinical trials are classified by the research objective created by the investigators.[12]
- In an observational study, the investigators observe the subjects and measure their outcomes. The researchers do not actively manage the study.[37]
- In an interventional study, the investigators give the research subjects an experimental drug, surgical procedure, use of a medical device, diagnostic or other intervention to compare the treated subjects with those receiving no treatment or the standard treatment. Then the researchers assess how the subjects' health changes.[37]
Trials are classified by their purpose. After approval for human research is granted to the trial sponsor, the U.S. Food and Drug Administration (FDA) organizes and monitors the results of trials according to type:[12]
- Prevention trials look for ways to prevent disease in people who have never had the disease or to prevent a disease from returning. These approaches may include drugs, vitamins or other micronutrients, vaccines, or lifestyle changes.
- Screening trials test for ways to identify certain diseases or health conditions.
- Diagnostic trials are conducted to find better tests or procedures for diagnosing a particular disease or condition.
- Treatment trials test experimental drugs, new combinations of drugs, or new approaches to surgery or radiation therapy.
- Quality of life trials (supportive care trials) evaluate how to improve comfort and quality of care for people with a chronic illness.
- Genetic trials are conducted to assess the prediction accuracy of genetic disorders making a person more or less likely to develop a disease.
- Epidemiological trials have the goal of identifying the general causes, patterns or control of diseases in large numbers of people.
- Compassionate use trials or expanded access trials provide partially tested, unapproved therapeutics to a small number of patients who have no other realistic options. Usually, this involves a disease for which no effective therapy has been approved, or a patient who has already failed all standard treatments and whose health is too compromised to qualify for participation in randomized clinical trials.[38] Usually, case-by-case approval must be granted by both the FDA and the pharmaceutical company for such exceptions.
- Fixed trials consider existing data only during the trial's design, do not modify the trial after it begins, and do not assess the results until the study is completed.
- Adaptive clinical trials use existing data to design the trial, and then use interim results to modify the trial as it proceeds. Modifications include dosage, sample size, drug undergoing trial, patient selection criteria and "cocktail" mix.[39] Adaptive trials often employ a Bayesian experimental design to assess the trial's progress. In some cases, trials have become an ongoing process that regularly adds and drops therapies and patient groups as more information is gained.[40] The aim is to more quickly identify drugs that have a therapeutic effect and to zero in on patient populations for whom the drug is appropriate.[41][42]
Clinical trials are conducted typically in four phases, with each phase using different numbers of subjects and having a different purpose to construct focus on identifying a specific effect.[12]
Phases[edit]
Clinical trials involving new drugs are commonly classified into five phases. Each phase of the drug approval process is treated as a separate clinical trial. The drug development process will normally proceed through phases I–IV over many years, frequently involving a decade or longer. If the drug successfully passes through phases I, II, and III, it will usually be approved by the national regulatory authority for use in the general population.[12] Phase IV trials are performed after the newly approved drug, diagnostic or device is marketed, providing assessment about risks, benefits, or best uses.[12]
Phase Aim Notes Phase 0 Pharmacodynamics and pharmacokinetics in humans Phase 0 trials are optional first-in-human trials. Single subtherapeutic doses of the study drug or treatment are given to a small number of subjects (typically 10 to 15) to gather preliminary data on the agent's pharmacodynamics (what the drug does to the body) and pharmacokinetics (what the body does to the drugs).[43] For a test drug, the trial documents the absorption, distribution, metabolization, and clearance (excretion) of the drug, and the drug's interactions within the body, to confirm that these appear to be as expected. Phase I Screening for safety Often are first-in-person trials. Testing within a small group of people (typically 20–80) to evaluate safety, determine safe dosage ranges, and identify side effects.[12] Phase II Establishing the preliminary efficacy of the drug in a "treatment group", usually against a placebo control group Phase II-a is specifically designed to assess dosing requirements (how much drug should be given),[12][44] while a Phase II-b trial is designed to determine efficacy (100–300 people),[1] assessing how well the drug works at the prescribed dose(s) to establish a therapeutic dose range and monitor for possible side effects.[44] Phase III Final confirmation of safety and efficacy Testing with large groups of people (typically 1,000–3,000) to confirm drug efficacy, evaluate its effectiveness, monitor side effects, compare it to commonly used treatments, and collect information that will allow it to be used safely.[12] Phase IV Safety studies during sales Postmarketing studies delineate risks, benefits, and optimal use. As such, they are ongoing during the drug's lifetime of active medical use.[12]
Trial design[edit]
A fundamental distinction in evidence-based practice is between observational studies and randomized controlled trials.[45] Types of observational studies in epidemiology, such as the cohort study and the case-control study, provide less compelling evidence than the randomized controlled trial.[45] In observational studies, the investigators retrospectively assess associations between the treatments given to participants and their health status, with potential for considerable errors in design and interpretation.[46]
A randomized controlled trial can provide compelling evidence that the study treatment causes an effect on human health.[45]
Some Phase II and most Phase III drug trials are designed as randomized, double-blind, and placebo-controlled.
- Randomized: Each study subject is randomly assigned to receive either the study treatment or a placebo.
- Blind: The subjects involved in the study do not know which study treatment they receive. If the study is double-blind, the researchers also do not know which treatment a subject receives. This intent is to prevent researchers from treating the two groups differently. A form of double-blind study called a "double-dummy" design allows additional insurance against bias. In this kind of study, all patients are given both placebo and active doses in alternating periods.
- Placebo-controlled: The use of a placebo (fake treatment) allows the researchers to isolate the effect of the study treatment from the placebo effect.
Clinical studies having small numbers of subjects may be "sponsored" by single researchers or a small group of researchers, and are designed to test simple questions or feasibility to expand the research for a more comprehensive randomized controlled trial.[47]
Clinical studies can be "sponsored" (financed and organized) by academic institutions, pharmaceutical companies, government entities and even private groups. Trials are conducted for new drugs, biotechnology, diagnostic assays or medical devices to determine their safety and efficacy prior to being submitted for regulatory review that would determine market approval.
Active control studies[edit]
In many cases, giving a placebo to a person suffering from a disease may be unethical.[48] To address this, it has become a common practice to conduct "active comparator" (also known as "active control") trials. In trials with an active control group, subjects are given either the experimental treatment or a previously approved treatment with known effectiveness.
Master protocol[edit]
In such studies multiple experimental treatments are tested in a single trial. Genetic testing enables researchers to group patients according to their genetic profile, deliver drugs based on that profile to that group and compare the results. Multiple companies can participate, each bringing a different drug. The first such approach targets squamous cell cancer, which includes varying genetic disruptions from patient to patient. Amgen, AstraZeneca and Pfizer are involved, the first time they have worked together in a late-stage trial. Patients whose genomic profiles do not match any of the trial drugs receive a drug designed to stimulate the immune system to attack cancer.[49]
Clinical trial protocol[edit]
A clinical trial protocol is a document used to define and manage the trial. It is prepared by a panel of experts. All study investigators are expected to strictly observe the protocol.
The protocol describes the scientific rationale, objective(s), design, methodology, statistical considerations and organization of the planned trial. Details of the trial are provided in documents referenced in the protocol, such as an investigator's brochure.
The protocol contains a precise study plan to assure safety and health of the trial subjects and to provide an exact template for trial conduct by investigators. This allows data to be combined across all investigators/sites. The protocol also informs the study administrators (often a contract research organization).
The format and content of clinical trial protocols sponsored by pharmaceutical, biotechnology or medical device companies in the United States, European Union, or Japan have been standardized to follow Good Clinical Practice guidance[50] issued by the International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH).[51] Regulatory authorities in Canada and Australia also follow ICH guidelines. Journals such as Trials, encourage investigators to publish their protocols.
Design features[edit]
Informed consent[edit]

Clinical trials recruit study subjects to sign a document representing their "informed consent".[52] The document includes details such as its purpose, duration, required procedures, risks, potential benefits, key contacts and institutional requirements.[53] The participant then decides whether to sign the document. The document is not a contract, as the participant can withdraw at any time without penalty.
Informed consent is a legal process in which a recruit is instructed about key facts before deciding whether to participate.[52] Researchers explain the details of the study in terms the subject can understand. The information is presented in the subject's native language. Generally, children cannot autonomously provide informed consent, but depending on their age and other factors, may be required to provide informed assent.
Statistical power[edit]
In any clinical trial, the number of subjects, also called the sample size, has a large impact on the ability to reliably detect and measure the effects of the intervention. This ability is described as its "power", which must be calculated before initiating a study to figure out if the study is worth its costs.[54] In general, a larger sample size increases the statistical power, also the cost.
The statistical power estimates the ability of a trial to detect a difference of a particular size (or larger) between the treatment and control groups. For example, a trial of a lipid-lowering drug versus placebo with 100 patients in each group might have a power of 0.90 to detect a difference between placebo and trial groups receiving dosage of 10 mg/dL or more, but only 0.70 to detect a difference of 6 mg/dL.
Placebo groups[edit]
Merely giving a treatment can have nonspecific effects. These are controlled for by the inclusion of patients who receive only a placebo. Subjects are assigned randomly without informing them to which group they belonged. Many trials are doubled-blinded so that researchers do not know to which group a subject is assigned.
Assigning a subject to a placebo group can pose an ethical problem if it violates his or her right to receive the best available treatment. The Declaration of Helsinki provides guidelines on this issue.
Duration[edit]

Clinical trials are only a small part of the research that goes into developing a new treatment. Potential drugs, for example, first have to be discovered, purified, characterized, and tested in labs (in cell and animal studies) before ever undergoing clinical trials. In all, about 1,000 potential drugs are tested before just one reaches the point of being tested in a clinical trial.[55] For example, a new cancer drug has, on average, six years of research behind it before it even makes it to clinical trials. But the major holdup in making new cancer drugs available is the time it takes to complete clinical trials themselves. On average, about eight years pass from the time a cancer drug enters clinical trials until it receives approval from regulatory agencies for sale to the public.[56] Drugs for other diseases have similar timelines.
Some reasons a clinical trial might last several years:
- For chronic conditions such as cancer, it takes months, if not years, to see if a cancer treatment has an effect on a patient.
- For drugs that are not expected to have a strong effect (meaning a large number of patients must be recruited to observe 'any' effect), recruiting enough patients to test the drug's effectiveness (i.e., getting statistical power) can take several years.
- Only certain people who have the target disease condition are eligible to take part in each clinical trial. Researchers who treat these particular patients must participate in the trial. Then they must identify the desirable patients and obtain consent from them or their families to take part in the trial.
A clinical trial might also include an extended post-study follow-up period from months to years for people who have participated in the trial, a so-called "extension phase", which aims to identify long-term impact of the treatment.[57]
The biggest barrier to completing studies is the shortage of people who take part. All drug and many device trials target a subset of the population, meaning not everyone can participate. Some drug trials require patients to have unusual combinations of disease characteristics. It is a challenge to find the appropriate patients and obtain their consent, especially when they may receive no direct benefit (because they are not paid, the study drug is not yet proven to work, or the patient may receive a placebo). In the case of cancer patients, fewer than 5% of adults with cancer will participate in drug trials. According to the Pharmaceutical Research and Manufacturers of America (PhRMA), about 400 cancer medicines were being tested in clinical trials in 2005. Not all of these will prove to be useful, but those that are may be delayed in getting approved because the number of participants is so low.[58]
For clinical trials involving potential for seasonal influences (such as airborne allergies, seasonal affective disorder, influenza, and skin diseases), the study may be done during a limited part of the year (such as spring for pollen allergies), when the drug can be tested.[59][60]
Clinical trials that do not involve a new drug usually have a much shorter duration. (Exceptions are epidemiological studies, such as the Nurses' Health Study).
Administration[edit]
Clinical trials designed by a local investigator, and (in the US) federally funded clinical trials, are almost always administered by the researcher who designed the study and applied for the grant. Small-scale device studies may be administered by the sponsoring company. Clinical trials of new drugs are usually administered by a contract research organization (CRO) hired by the sponsoring company. The sponsor provides the drug and medical oversight. A CRO is contracted to perform all the administrative work on a clinical trial. For Phases II–IV the CRO recruits participating researchers, trains them, provides them with supplies, coordinates study administration and data collection, sets up meetings, monitors the sites for compliance with the clinical protocol, and ensures the sponsor receives data from every site. Specialist site management organizations can also be hired to coordinate with the CRO to ensure rapid IRB/IEC approval and faster site initiation and patient recruitment. Phase I clinical trials of new medicines are often conducted in a specialist clinical trial clinic, with dedicated pharmacologists, where the subjects can be observed by full-time staff. These clinics are often run by a CRO which specialises in these studies.
At a participating site, one or more research assistants (often nurses) do most of the work in conducting the clinical trial. The research assistant's job can include some or all of the following: providing the local institutional review board (IRB) with the documentation necessary to obtain its permission to conduct the study, assisting with study start-up, identifying eligible patients, obtaining consent from them or their families, administering study treatment(s), collecting and statistically analyzing data, maintaining and updating data files during followup, and communicating with the IRB, as well as the sponsor and CRO.
Quality[edit]
In the context of a clinical trial, quality typically refers to the absence of errors which can impact decision making, both during the conduct of the trial and in use of the trial results.[61]
Marketing[edit]
An Interactional Justice Model may be used to test the effects of willingness to talk with a doctor about clinical trial enrollment.[62] Results found that potential clinical trial candidates were less likely to enroll in clinical trials if the patient is more willing to talk with their doctor. The reasoning behind this discovery may be patients are happy with their current care. Another reason for the negative relationship between perceived fairness and clinical trial enrollment is the lack of independence from the care provider. Results found that there is a positive relationship between a lack of willingness to talk with their doctor and clinical trial enrollment. Lack of willingness to talk about clinical trials with current care providers may be due to patients' independence from the doctor. Patients who are less likely to talk about clinical trials are more willing to use other sources of information to gain a better insight of alternative treatments. Clinical trial enrollment should be motivated to utilize websites and television advertising to inform the public about clinical trial enrollment.
Information technology[edit]
The last decade has seen a proliferation of information technology use in the planning and conduct of clinical trials. Clinical trial management systems are often used by research sponsors or CROs to help plan and manage the operational aspects of a clinical trial, particularly with respect to investigational sites. Advanced analytics for identifying researchers and research sites with expertise in a given area utilize public and private information about ongoing research.[63] Web-based electronic data capture (EDC) and clinical data management systems are used in a majority of clinical trials[64] to collect case report data from sites, manage its quality and prepare it for analysis. Interactive voice response systems are used by sites to register the enrollment of patients using a phone and to allocate patients to a particular treatment arm (although phones are being increasingly replaced with web-based (IWRS) tools which are sometimes part of the EDC system). While patient-reported outcome were often paper based in the past, measurements are increasingly being collected using web portals or hand-held ePRO (or eDiary) devices, sometimes wireless.[65] Statistical software is used to analyze the collected data and prepare them for regulatory submission. Access to many of these applications are increasingly aggregated in web-based clinical trial portals. In 2011, the FDA approved a Phase I trial that used telemonitoring, also known as remote patient monitoring, to collect biometric data in patients' homes and transmit it electronically to the trial database. This technology provides many more data points and is far more convenient for patients, because they have fewer visits to trial sites.
Analysis[edit]
A clinical trial produces data that could reveal quantitative differences between two or more interventions; statistical analyses are used to determine whether such differences are true, result from chance, or are the same as no treatment (placebo).[66][67] Data from a clinical trial accumulate gradually over the trial duration, extending from months to years.[52] Accordingly, results for participants recruited early in the study become available for analysis while subjects are still being assigned to treatment groups in the trial. Early analysis may allow the emerging evidence to assist decisions about whether to stop the study, or to reassign participants to the more successful segment of the trial.[66] Investigators may also want to stop a trial when data analysis shows no treatment effect.[67]
Ethical aspects[edit]
Clinical trials are closely supervised by appropriate regulatory authorities. All studies involving a medical or therapeutic intervention on patients must be approved by a supervising ethics committee before permission is granted to run the trial. The local ethics committee has discretion on how it will supervise noninterventional studies (observational studies or those using already collected data). In the US, this body is called the Institutional Review Board (IRB); in the EU, they are called Ethics committees. Most IRBs are located at the local investigator's hospital or institution, but some sponsors allow the use of a central (independent/for profit) IRB for investigators who work at smaller institutions.
Чтобы быть этичными, исследователи должны получить полное и информированное согласие участвующих людей. (Одна из основных функций IRB — обеспечить адекватное информирование потенциальных пациентов о клиническом исследовании.) Если пациент не может дать согласие от себя, исследователи могут запросить согласие у законного представителя пациента. Кроме того, участники клинического исследования должны быть проинформированы о том, что они могут выйти из клинического исследования в любое время без каких-либо неблагоприятных действий против них. [68] В Калифорнии штат отдает приоритет лицам, которые могут выступать в качестве законных представителей. [69]
В некоторых регионах США местный IRB должен сертифицировать исследователей и их сотрудников, прежде чем они смогут проводить клинические испытания. Они должны понимать федеральный закон о конфиденциальности пациентов ( HIPAA ) и надлежащую клиническую практику. Международная конференция по гармонизации руководящих принципов надлежащей клинической практики представляет собой набор стандартов, используемых на международном уровне для проведения клинических исследований. Руководящие принципы направлены на обеспечение «защиты прав, безопасности и благополучия субъектов испытаний».
Понятие информированного согласия участвующих людей существует во многих странах, но его точное определение может различаться.
Информированное согласие, очевидно, является «необходимым» условием этического поведения, но не «обеспечивает» этическое поведение. В ходе испытаний сострадательного использования последнее становится особенно сложной проблемой. Конечная цель — служить сообществу пациентов или будущих пациентов наилучшим и наиболее ответственным образом. См. также Расширенный доступ . Однако может быть сложно превратить эту цель в четко определенную, количественную целевую функцию. Однако в некоторых случаях это можно сделать, например, для вопросов о том, когда прекратить последовательные обработки (см. Алгоритм шансов ), и тогда количественные методы могут сыграть важную роль.
Дополнительные этические проблемы возникают при проведении клинических исследований на детях ( педиатрия ), а также в чрезвычайных или эпидемических ситуациях. [70] [71]
Этический баланс прав нескольких заинтересованных сторон может быть трудным. Например, если испытания лекарства терпят неудачу, спонсоры могут быть обязаны немедленно сообщить об этом нынешним и потенциальным инвесторам, а это означает, что и исследовательский персонал, и зарегистрированные участники могут сначала узнать об окончании испытания из публичных деловых новостей . [72]
интересов и неблагоприятные Конфликты исследования
В ответ на конкретные случаи, когда неблагоприятные данные исследований, спонсируемых фармацевтическими компаниями, не публиковались, Фармацевтические исследования и производители Америки опубликовали новые рекомендации, призывающие компании сообщать обо всех результатах и ограничивать финансовое участие исследователей в фармацевтических компаниях. [73] Конгресс США подписал закон, который требует, чтобы клинические испытания фазы II и III регистрировались спонсором на веб-сайте Clinicaltrials.gov, созданном Национальными институтами здравоохранения . [74]
Исследователи лекарств, не работающие напрямую в фармацевтических компаниях, часто ищут гранты у производителей, а производители часто обращаются к академическим исследователям для проведения исследований в сетях университетов и их больниц, например, для трансляционных исследований рака. Аналогичным образом, конкуренция за постоянные академические должности, государственные гранты и престиж создают конфликты интересов среди ученых-академиков. [75] Согласно одному исследованию, примерно 75% статей, отозванных по причинам, связанным с неправомерным поведением, не имеют заявленной финансовой поддержки со стороны отрасли. [76] испытания посева . Особые споры вызывают [77]
В Соединенных Штатах все клинические испытания, представленные в FDA в рамках процесса утверждения лекарств, проходят независимую оценку клинических экспертов Управления по контролю за продуктами и лекарствами. [78] включая проверки сбора первичных данных в отдельных центрах клинических исследований. [79]
В 2001 году редакторы 12 крупных журналов опубликовали совместную редакционную статью, опубликованную в каждом журнале, о контроле за клиническими исследованиями, проводимыми спонсорами, в частности, касаясь использования контрактов, которые позволяют спонсорам просматривать исследования до публикации и воздерживаться от публикации. Они усилили редакционные ограничения, чтобы противостоять этому эффекту. В редакционной статье отмечалось, что к 2000 году контрактные исследовательские организации получили 60% грантов от фармацевтических компаний США. Исследователям может быть запрещено участвовать в разработке дизайна исследования, получать доступ к необработанным данным и интерпретировать результаты. [80]
Несмотря на явные рекомендации заинтересованных сторон о мерах по улучшению стандартов медицинских исследований, спонсируемых промышленностью, [81] В 2013 году Тоэн предупредил о сохранении пробела в достоверности выводов, вытекающих из клинических испытаний, финансируемых промышленностью, и призвал обеспечить строгое соблюдение этических стандартов в промышленном сотрудничестве с научными кругами, чтобы избежать дальнейшей эрозии доверия общественности. [82] Вопросы, требующие внимания в этом отношении, включают потенциальную систематическую ошибку наблюдения, продолжительность времени наблюдения для поддерживающих исследований, выбор популяции пациентов, факторы, влияющие на реакцию плацебо, и источники финансирования. [83] [84] [85]
здравоохранения кризиса Во время общественного
Проведение клинических испытаний вакцин во время эпидемий и пандемий сопряжено с этическими проблемами. Для болезней с высоким уровнем смертности, таких как Эбола, отнесение людей к группе плацебо или контрольной группе может рассматриваться как смертный приговор. В ответ на этические проблемы, связанные с клиническими исследованиями во время эпидемий, Национальная академия медицины подготовила отчет, в котором указаны семь этических и научных соображений. Эти соображения таковы: [86]
- Научная ценность
- Социальная ценность
- Уважение к личности
- Участие сообщества
- Забота о благополучии и интересах участников
- Баланс между выгодой и риском
- Пост-испытательный доступ к проверенным методам лечения, которые были закрыты во время исследования
Беременные женщины и дети [ править ]
Беременных женщин и детей обычно исключают из клинических исследований как уязвимые группы населения, хотя данные, подтверждающие их исключение, не являются надежными. Исключая их из клинических испытаний, информация о безопасности и эффективности терапии для этих групп населения часто отсутствует. На заре эпидемии ВИЧ/СПИДа один ученый заметил, что, исключая эти группы из потенциально жизненно важного лечения, они были «защищены до смерти». Такие проекты, как «Этика исследований вакцин, эпидемий и новых технологий» (ПРЕВЕНТ), выступают за этическое включение беременных женщин в испытания вакцин. Включение детей в клинические испытания имеет дополнительные моральные соображения, поскольку детям не хватает самостоятельности в принятии решений. В прошлом испытания подвергались критике за использование госпитализированных детей или сирот; эти этические проблемы фактически остановили будущие исследования. Стремясь поддерживать эффективную педиатрическую помощь, некоторые европейские страны и США проводят политику, направленную на то, чтобы побудить или заставить фармацевтические компании проводить педиатрические исследования. Международные руководства рекомендуют проводить этические педиатрические исследования, ограничивая вред, учитывая различные риски и принимая во внимание сложности педиатрической помощи. [86]
Безопасность [ править ]
Ответственность за безопасность участников клинического исследования распределяется между спонсором, местными исследователями (если они отличаются от спонсора), различными IRB, которые контролируют исследование, и (в некоторых случаях, если в исследовании используется коммерческий препарат) или устройство), регулирующий орган страны, где будет продаваться препарат или устройство.
Для обеспечения безопасности участников исследования часто используется систематический одновременный анализ безопасности. Проведение и текущий обзор должны быть пропорциональны риску, связанному с испытанием. Обычно эту роль выполняет Комитет по данным и безопасности , назначенный извне наблюдатель за медицинской безопасностью. [87] Независимый специалист по безопасности или, в случае небольших исследований или исследований с низким уровнем риска, главный исследователь. [88]
Из соображений безопасности многие клинические испытания лекарств [89] предназначены для исключения женщин детородного возраста, беременных женщин или женщин, которые забеременели во время исследования. В некоторых случаях партнеры-мужчины этих женщин также исключаются или обязаны принимать меры контроля над рождаемостью.
Спонсор [ править ]
На протяжении всего клинического исследования спонсор несет ответственность за точное информирование местных исследователей об истинных исторических данных о безопасности препарата, устройства или других медицинских методов лечения, подлежащих тестированию, а также о любых потенциальных взаимодействиях исследуемого препарата(ов) с уже одобренными препаратами. методы лечения. Это позволяет местным исследователям принять обоснованное решение о том, участвовать ли в исследовании или нет. Спонсор также несет ответственность за мониторинг результатов исследования по мере их поступления из различных учреждений по мере проведения исследования. В более крупных клинических исследованиях спонсор будет пользоваться услугами комитета по мониторингу данных (DMC, известного в США как совет по мониторингу безопасности данных). Эта независимая группа клиницистов и статистиков периодически встречается для рассмотрения открытых данных, полученных спонсором на данный момент. DMC имеет право рекомендовать прекращение исследования на основании своего обзора, например, если исследуемый препарат вызывает больше смертей, чем стандартное лечение, или кажется, что он вызывает неожиданные и серьезные последствия, связанные с исследованием. неблагоприятные события . Спонсор несет ответственность за сбор отчетов о нежелательных явлениях от всех исследователей, участвующих в исследовании, а также за информирование всех исследователей о заключении спонсора относительно того, были ли эти нежелательные явления связаны или не связаны с исследуемым лечением.
Спонсор и местные исследователи несут совместную ответственность за составление информированного согласия для конкретного участка , которое точно информирует потенциальных субъектов об истинных рисках и потенциальных преимуществах участия в исследовании, в то же время представляя материал как можно более кратко и на обычном языке. В правилах FDA говорится, что участие в клинических исследованиях является добровольным, при этом субъект имеет право не участвовать или прекратить участие в любое время. [90]
Местные следователи [ править ]
В исследовании используется этический принцип primum non-nocere («прежде всего, не навреди»), и если исследователь считает, что исследуемое лечение может нанести вред субъектам исследования, он может прекратить участие в исследовании в любое время. С другой стороны, следователи часто имеют финансовую заинтересованность в привлечении субъектов и могут действовать неэтично, чтобы добиться и сохранить их участие.
Местные исследователи несут ответственность за проведение исследования в соответствии с протоколом исследования и контроль за персоналом, проводящим исследование, на протяжении всего исследования. Местный исследователь или его/ее исследовательский персонал также несут ответственность за обеспечение понимания потенциальными участниками исследования рисков и потенциальных преимуществ участия в исследовании. Другими словами, они (или их законные представители) должны дать действительно осознанное согласие.
Местные исследователи несут ответственность за рассмотрение всех отчетов о нежелательных явлениях, отправленных спонсором. Эти отчеты о нежелательных явлениях содержат мнения как исследователя (в месте, где произошло нежелательное явление), так и спонсора относительно связи нежелательного явления с исследуемым лечением. Местные исследователи также несут ответственность за вынесение независимого заключения по этим отчетам и оперативное информирование местного IRB обо всех серьезных и связанных с исследуемым лечением нежелательных явлениях.
Если спонсором является местный исследователь, формальных отчетов о нежелательных явлениях может не быть, но персонал исследования во всех местах несет ответственность за информирование исследователя-координатора о любых неожиданных событиях. Местный исследователь несет ответственность за предоставление правдивой информации местному IRB во всех сообщениях, касающихся исследования.
Институциональные наблюдательные советы (IRB) [ править ]
одобрение Институционального наблюдательного совета (IRB) или Независимого комитета по этике Прежде чем начать любое исследование, кроме самого неформального, необходимо (IEC). В коммерческих клинических исследованиях протокол исследования не утверждается IRB до того, как спонсор наберет места для проведения исследования. Однако протокол и процедуры исследования были адаптированы в соответствии с общими требованиями к подаче заявок в IRB. В этом случае и при отсутствии независимого спонсора каждый местный исследователь представляет протокол исследования, согласие(я), формы сбора данных и подтверждающую документацию в местный ЭСО. Университеты и большинство больниц имеют собственные IRB. Другие исследователи (например, в поликлиниках) используют независимые IRB.
IRB тщательно проверяет исследование как на предмет медицинской безопасности, так и на предмет защиты пациентов, участвующих в исследовании, прежде чем разрешить исследователю начать исследование. Это может потребовать внесения изменений в процедуры исследования или в объяснения, данные пациенту. Требуемый ежегодный отчет исследователя о «постоянном обзоре» информирует IRB о ходе исследования и любой новой информации о безопасности, связанной с исследованием.
Регулирующие органы [ править ]
В США FDA может проверять файлы местных исследователей после того, как они завершили участие в исследовании, чтобы проверить, правильно ли они следовали процедурам исследования. Такая проверка может быть выборочной или поводовой (поскольку следователь подозревается в подтасовке данных). Уклонение от проверки является для следователей стимулом следовать процедурам исследования. «Покрываемое клиническое исследование» относится к исследованию, поданному в FDA как часть маркетинговой заявки (например, как часть NDA или 510 (k) ), в отношении которого FDA может потребовать раскрытия финансового интереса клинического исследователя. в результате исследования. Например, заявитель должен раскрыть, владеет ли исследователь долей в спонсоре или имущественной долей в исследуемом продукте. FDA определяет контролируемое исследование как «... любое исследование лекарственного средства, биологического продукта или устройства на людях, представленное в маркетинговой заявке или ходатайстве о реклассификации, на которое заявитель или FDA опирается для установления эффективности продукта (включая исследования, которые показывают эквивалентность эффективного продукта) или любое исследование, в котором один исследователь вносит значительный вклад в демонстрацию безопасности». [91]
Альтернативно, многие американские фармацевтические компании перенесли некоторые клинические испытания за границу. Преимущества проведения исследований за рубежом включают более низкие затраты (в некоторых странах) и возможность проводить более крупные исследования в более короткие сроки, тогда как потенциальный недостаток заключается в более низком качестве управления исследованиями. [92] В разных странах действуют разные нормативные требования и возможности правоприменения. По оценкам, 40% всех клинических испытаний сейчас проводятся в Азии, Восточной Европе, Центральной и Южной Америке. «В этих странах нет обязательной системы регистрации клинических испытаний, и многие из них не следуют европейским директивам в своей деятельности», — говорит Джейкоб Сийтсма из нидерландской WEMOS, правозащитной медицинской организации, отслеживающей клинические испытания в развивающихся странах. [93]
Начиная с 1980-х годов, гармонизация протоколов клинических испытаний оказалась осуществимой во всех странах Европейского Союза. В то же время координация между Европой, Японией и Соединенными Штатами привела к совместной инициативе регуляторной отрасли по международной гармонизации, названной в честь 1990 года Международной конференцией по гармонизации технических требований к регистрации фармацевтических препаратов для использования человеком (ICH). [94] В настоящее время большинство программ клинических испытаний следуют рекомендациям ICH, направленным на «обеспечение того, чтобы качественные, безопасные и эффективные лекарства разрабатывались и регистрировались наиболее эффективным и экономически выгодным способом. Эта деятельность осуществляется в интересах потребителей и общественного здравоохранения». предотвратить ненужное дублирование клинических испытаний на людях и свести к минимуму использование испытаний на животных без ущерба для нормативных обязательств по безопасности и эффективности». [95]
Агрегация данных по безопасности в ходе клинической разработки [ править ]
Обобщение данных о безопасности клинических испытаний во время разработки лекарств важно, поскольку исследования обычно проводятся с целью определить, насколько хорошо действует препарат. Данные о безопасности, собранные и обобщенные в ходе многочисленных исследований по мере разработки препарата, позволяют спонсору, исследователям и регулирующим органам отслеживать совокупный профиль безопасности экспериментальных лекарств по мере их разработки. Ценность оценки совокупных данных о безопасности заключается в том, что: а) решения, основанные на совокупной оценке безопасности во время разработки лекарственного средства, могут быть приняты на протяжении всей разработки лекарственного средства и б) это дает спонсору и регулирующим органам хорошую подготовку для оценки безопасности лекарственного средства после его утверждения. . [96] [97] [98] [99] [100]
Экономика [ править ]
Стоимость клинических исследований варьируется в зависимости от фазы исследования, типа исследования и изучаемого заболевания. Исследование клинических испытаний, проведенных в США с 2004 по 2012 год, показало, что средняя стоимость исследований фазы I составляет от 1,4 до 6,6 миллионов долларов, в зависимости от типа заболевания. Стоимость испытаний фазы II варьировалась от 7 до 20 миллионов долларов, а испытаний фазы III — от 11 до 53 миллионов долларов. [101]
Спонсор [ править ]
Стоимость исследования зависит от многих факторов, особенно от количества центров, проводящих исследование, количества участвующих пациентов и того, одобрено ли уже исследуемое лечение для медицинского использования.
Расходы, понесенные фармацевтической компанией при проведении клинических исследований фазы III или IV, могут включать, среди прочего:
- производство оцениваемого препарата(ов) или устройства(ий)
- зарплата дизайнеров и администраторов исследования
- выплаты контрактной исследовательской организации, организации по управлению объектом (если она используется) и любым внешним консультантам
- выплаты местным исследователям и их сотрудникам за время и усилия по набору испытуемых и сбору данных для спонсора
- стоимость учебных материалов и расходы на их доставку
- общение с местными исследователями, включая мониторинг на месте со стороны CRO до и (в некоторых случаях) несколько раз во время исследования
- одно или несколько учебных совещаний для следователей
- расходы, понесенные местными исследователями, такие как сборы за аптеку, сборы IRB и почтовые расходы.
- любые выплаты субъектам, участвующим в исследовании
- расходы на лечение испытуемого, у которого развилось заболевание, вызванное исследуемым препаратом
Эти расходы производятся в течение нескольких лет.
В США спонсоры могут получить 50-процентную налоговую льготу за клинические испытания лекарств, разрабатываемых для лечения орфанных заболеваний . [102] Национальные агентства здравоохранения, такие как Национальные институты здравоохранения США , предлагают гранты исследователям, которые разрабатывают клинические испытания, которые пытаются ответить на исследовательские вопросы, интересующие агентство. В этих случаях исследователь, который выдает грант и проводит исследование, выступает в качестве спонсора и координирует сбор данных с любых других сайтов. Этим другим сайтам может быть оплачено или не оплачено участие в исследовании, в зависимости от суммы гранта и ожидаемых от них усилий. Использование интернет-ресурсов может в некоторых случаях снизить экономическое бремя. [103]
Следователи [ править ]
Исследователи часто получают вознаграждение за свою работу в клинических исследованиях. Эти суммы могут быть небольшими, покрывая лишь частичную заработную плату научных сотрудников и стоимость любых расходных материалов (обычно в случае исследований национальных агентств здравоохранения), или быть значительными и включать «накладные расходы», которые позволяют исследователю платить исследовательскому персоналу в периоды времени. между клиническими исследованиями. [ нужна ссылка ]
Предметы [ править ]
Участники I фазы испытаний лекарств не получают от участия никакой прямой пользы для здоровья. Обычно им платят за потраченное время, причем выплаты регулируются и не связаны с каким-либо риском. Мотивация здоровых добровольцев не ограничивается финансовым вознаграждением и может включать в себя и другие мотивы, такие как вклад в науку и другие. [104] На более поздних стадиях испытаний испытуемым могут не платить за обеспечение их мотивации к участию с потенциальной пользой для здоровья или вкладом в медицинские знания. Небольшие выплаты могут производиться на расходы, связанные с исследованием, такие как проезд, или в качестве компенсации за время, потраченное на предоставление последующей информации о своем здоровье после окончания пробного лечения.
Набор участников и участие [ править ]

Для испытаний лекарств фазы 0 и фазы I требуются здоровые добровольцы. В большинстве других клинических испытаний участвуют пациенты с определенным заболеванием или состоянием здоровья. Разнообразие, наблюдаемое в обществе, должно быть отражено в клинических исследованиях посредством надлежащего включения этнических меньшинств . [105] Набор пациентов или набор участников играет значительную роль в деятельности и обязанностях центров, проводящих клинические исследования. [106]
Все добровольцы, рассматриваемые для участия в исследовании, обязаны пройти медицинский осмотр. Требования различаются в зависимости от потребностей исследования, но обычно добровольцев проверяют в медицинской лаборатории на: [107]
- Измерение электрической активности сердца (ЭКГ)
- Измерение артериального давления, частоты сердечных сокращений и температуры тела
- Забор крови
- Забор мочи
- Измерение веса и роста
- Тестирование на злоупотребление наркотиками
- Тест на беременность
Было замечено, что участники клинических испытаний непропорционально белые. [108] [109] Зачастую меньшинства не информируют о клинических испытаниях. [110] Один недавний систематический обзор литературы показал, что раса/этническая принадлежность, а также пол не были широко представлены и иногда даже не отслеживались в качестве участников в большом количестве клинических исследований по лечению потери слуха у взрослых. [111] Это может снизить достоверность результатов в отношении небелых пациентов. [112] не представляя адекватно большую часть населения .
Поиск испытаний [ править ]
В зависимости от требуемого типа участников спонсоры клинических исследований или контрактные исследовательские организации, работающие от их имени, стараются найти места с квалифицированным персоналом, а также доступ к пациентам, которые могли бы принять участие в исследовании. Работая с этими сайтами, они могут использовать различные стратегии набора персонала, включая базы данных пациентов, рекламу в газетах и радио, листовки, плакаты в местах, куда пациенты могут пойти (например, в кабинеты врачей), а также личный набор пациентов исследователями.
Добровольцы с конкретными заболеваниями или заболеваниями имеют дополнительные онлайн-ресурсы, которые помогут им найти клинические испытания. Например, Fox Trial Finder объединяет исследования болезни Паркинсона по всему миру с добровольцами, имеющими определенный набор критериев, таких как местоположение, возраст и симптомы. [113] Существуют и другие службы, специализирующиеся на конкретных заболеваниях, позволяющие добровольцам найти исследования, связанные с их состоянием. [114] Добровольцы могут искать исследования непосредственно на сайте ClinicalTrials.gov, используя реестр, который ведется Национальными институтами здравоохранения США и Национальной медицинской библиотекой . Существует также программное обеспечение, которое позволяет клиницистам находить варианты испытаний для отдельного пациента на основе таких данных, как геномные данные. [115]
Исследования [ править ]

Модель поиска и обработки информации о рисках (RISP) анализирует социальные последствия, которые влияют на отношение и принятие решений, касающихся клинических испытаний. [116] Люди, которые более заинтересованы в лечении, проводимом в рамках клинических исследований, с большей вероятностью будут искать информацию о клинических исследованиях. Больные раком сообщали о более оптимистичном отношении к клиническим испытаниям, чем население в целом. Более оптимистичный взгляд на клинические испытания также приводит к большей вероятности участия в них. [116]
Соответствие [ править ]
Сопоставление включает систематическое сравнение клинической и демографической информации пациента с критериями отбора различных исследований. Методы включают в себя:
- Руководство: Медицинские работники или координаторы клинических исследований вручную просматривают записи пациентов и доступные критерии исследования, чтобы выявить потенциальные совпадения. [117] Это также может включать ручной поиск в базах данных клинических исследований. [118]
- Электронные медицинские карты (ЭМК). Некоторые системы интегрируются с электронными медицинскими документами, чтобы автоматически отмечать пациентов, которые могут иметь право на участие в исследованиях, на основе их медицинских данных. Эти системы могут использовать методы машинного обучения , искусственного интеллекта или точной медицины для более эффективного подбора пациентов для участия в исследованиях. [119] Эти методы сталкиваются с проблемой преодоления ограничений записей EHR, таких как пропуски и ошибки регистрации.
- Услуги, ориентированные непосредственно на пациентов. Ресурсы специализируются на оказании помощи пациентам в поиске клинических испытаний через онлайн-платформы, горячие линии и персонализированную поддержку. [117] [118] [120]
Децентрализованные испытания [ править ]
Хотя исследования обычно проводятся в крупных медицинских центрах, некоторые участники исключаются из них из-за расстояния и расходов, необходимых для поездки, что приводит к трудностям, невыгодному состоянию и неравенству для участников, особенно тех, кто проживает в сельских и недостаточно обслуживаемых общинах. В 21 веке предпринимаются усилия по сбору информации в доме участника, и эта возможность улучшается благодаря телемедицине и портативным технологиям . [121]
См. также [ править ]
Ссылки [ править ]
- ↑ Перейти обратно: Перейти обратно: а б с «Клинические исследования НИЗ и вы: основы» . Национальные институты здравоохранения США. 3 октября 2022 года. Архивировано из оригинала 22 апреля 2020 года . Проверено 7 ноября 2022 г.
- ^ «Клинические испытания» (PDF) . Фонд Билла и Мелинды Гейтс. Архивировано (PDF) из оригинала 12 января 2017 года . Проверено 1 января 2014 г.
- ^ Димаси, Джозеф А; Грабовски, Генри Дж.; Хансен, Рональд В. (2016). «Инновации в фармацевтической промышленности: новые оценки затрат на НИОКР». Журнал экономики здравоохранения . 47 : 20–33. дои : 10.1016/j.jhealeco.2016.01.012 . hdl : 10161/12742 . ПМИД 26928437 .
- ^ «Процесс разработки лекарств. Шаг 3: Клинические исследования» . Управление по контролю за продуктами и лекарствами США. 4 января 2018 года . Проверено 9 марта 2024 г.
- ^ «Сколько времени нужно новому лекарству, чтобы пройти клинические испытания» . Исследования рака, Великобритания. 1 февраля 2022 г. Проверено 9 марта 2024 г.
- ^ Эмануэль Э.Дж. (9 сентября 2015 г.). «Решение проблемы цен на лекарства» . Нью-Йорк Таймс . Архивировано из оригинала 5 декабря 2022 года . Проверено 26 февраля 2017 г.
Из лекарств, начатых в клинических испытаниях на людях, только 10 процентов получили одобрение FDA. ...
- ^ Страница FDA последнее обновление 25 апреля 2014 г. Процесс проверки лекарств FDA: продолжение. Архивировано 23 апреля 2019 г. на Wayback Machine.
- ^ ФРМА. , февраль 2007 г. Открытие и разработка лекарств Архивировано 10 апреля 2023 г. в Wayback Machine.
- ^ Руководство Merck. Последний полный обзор/пересмотр: октябрь 2013 г., автор Дэниел А. Хусар, доктор философии. Обзор лекарств, отпускаемых без рецепта. Архивировано 4 февраля 2015 г. в Wayback Machine.
- ^ Аворн Дж. (2004). Мощные лекарства , стр. 129–33. Альфред А. Кнопф.
- ^ Ван Сполл Х.Г., Торен А., Кисс А., Фаулер Р.А. (март 2007 г.). «Критерии отбора рандомизированных контролируемых исследований, опубликованных в влиятельных общемедицинских журналах: систематический выборочный обзор». ДЖАМА . 297 (11): 1233–40. дои : 10.1001/jama.297.11.1233 . ПМИД 17374817 .
- ↑ Перейти обратно: Перейти обратно: а б с д и ж г час я дж «Каковы различные типы клинических исследований?» . Управление по контролю за продуктами и лекарствами США. 2019. Архивировано из оригинала 12 июня 2019 года . Проверено 24 мая 2019 г.
- ^ Регулирующим органом в США является Управление по контролю за продуктами и лекарствами ; в Канаде – Министерство здравоохранения Канады ; в Европейском Союзе – Европейское агентство по лекарственным средствам ; а в Японии – Министерство здравоохранения, труда и социального обеспечения.
- ^ «Медицинские изделия, предпродажные клинические исследования для исключения исследуемых устройств» . Управление по контролю за продуктами и лекарствами США. 17 марта 2017 г. Архивировано из оригинала 1 ноября 2017 г. Проверено 2 октября 2017 г.
- ^ Ледерле Ф.А., Фрейшлаг Х.А., Кириакидес Т.К., Падберг Ф.Т., Мацумура Дж.С., Колер Т.Р., Лин П.Х., Жан-Клод Ж.М., Цикрит Д.Ф., Суонсон К.М., Педуцци П.Н. (октябрь 2009 г.). «Результаты эндоваскулярного и открытого лечения аневризмы брюшной аорты: рандомизированное исследование» . ДЖАМА . 302 (14): 1535–42. дои : 10.1001/jama.2009.1426 . ПМИД 19826022 .
- ^ Липп А., Шоу С., Главинд К. (декабрь 2014 г.). «Механические устройства для лечения недержания мочи у женщин» . Кокрановская база данных систематических обзоров . 2014 (12): CD001756. дои : 10.1002/14651858.CD001756.pub6 . ПМК 7061494 . ПМИД 25517397 .
- ^ Фаррохьяр Ф., Караниколас П.Дж., Тома А., Симунович М., Бхандари М., Деверо П.Дж., Анвари М., Адили А., Гаятт Г. (март 2010 г.). «Рандомизированные контролируемые исследования хирургических вмешательств». Анналы хирургии . 251 (3): 409–16. дои : 10.1097/SLA.0b013e3181cf863d . ПМИД 20142732 . S2CID 17084906 .
- ^ Метвалли, Мостафа; Рэйбоулд, Грейс; Чеонг, Ин С; Хорн, Эндрю В. (29 января 2020 г.). Кокрейновская группа по гинекологии и фертильности (ред.). «Хирургическое лечение миомы матки при бесплодии» . Кокрейновская база данных систематических обзоров . 2020 (1): CD003857. дои : 10.1002/14651858.CD003857.pub4 . ПМК 6989141 . ПМИД 31995657 .
- ^ Цао А.М., Кокс М.Р., Эслик Г.Д. (март 2016 г.). «План исследования в доказательной хирургии: какова роль исследований случай-контроль?» . Всемирный методологический журнал . 6 (1): 101–4. дои : 10.5662/wjm.v6.i1.101 . ПМЦ 4804244 . ПМИД 27019801 .
- ^ «Пакет общественной информации (PIP): Как принять участие в исследованиях Национальной службы здравоохранения, общественного здравоохранения и социальной помощи» . Национальный институт исследований в области здравоохранения и ухода . Проверено 3 января 2024 г.
- ^ «Информационные заметки для исследователей – участие общественности в исследованиях Национальной службы здравоохранения, здравоохранения и социальной помощи» . Национальный институт исследований в области здравоохранения и ухода . Проверено 3 января 2024 г.
- ↑ Перейти обратно: Перейти обратно: а б с д Мейнерт КЛ, Тонасия С (1986). Клинические исследования: дизайн, проведение и анализ . Издательство Оксфордского университета, США. п. 3. ISBN 978-0-19-503568-1 . Архивировано из оригинала 15 апреля 2023 года . Проверено 5 июня 2020 г.
- ^ Саймон, Харви Б. (2002). Руководство Гарвардской медицинской школы по мужскому здоровью . Нью-Йорк: Свободная пресса . п. 31 . ISBN 978-0-684-87181-3 .
- ^ Браун, Стивен Р. (2003). Цинга: как хирург, моряк и джентльмен разгадали величайшую медицинскую загадку эпохи парусного спорта . Нью-Йорк, штат Нью-Йорк: Пресса Св. Мартина. ISBN 0-312-31391-8
- ^ Роджерс, Эверетт М. (1995). Распространение инноваций . Нью-Йорк, штат Нью-Йорк: Свободная пресса. ISBN 0-7432-2209-1 . п. 7.
- ^ Карлайл, Родни (2004). Scientific American Inventions and Discoveries , John Wiley & Songs, Inc., Нью-Джерси. п. 393. ISBN 0-471-24410-4 .
- ^ «Джеймс Линд: Трактат о цинге (1754 г.)» . 2001. Архивировано из оригинала 1 октября 2018 года . Проверено 9 сентября 2007 г.
- ^ «О Дне клинических исследований» . День клинических испытаний . Архивировано из оригинала 5 февраля 2023 года . Проверено 7 апреля 2023 г.
- ^ Грин С., Кроули Дж., Бенедетти Дж., Смит А. (30 июля 2002 г.). Клинические исследования в онкологии, второе издание . ЦРК Пресс. стр. 1–. ISBN 978-1-4200-3530-8 .
- ^ Гад СК (17 июня 2009 г.). Справочник по клиническим исследованиям . Джон Уайли и сыновья. стр. 118–. ISBN 978-0-470-46635-3 .
- ^ О'Рурк МФ (февраль 1992 г.). «Фредерик Акбар Магомед» . Гипертония . 19 (2): 212–7. дои : 10.1161/01.HYP.19.2.212 . ПМИД 1737655 .
- ^ Кресвелл, JW (2008). Образовательные исследования: планирование, проведение и оценка количественных и качественных исследований (3-е). Река Аппер-Седл, Нью-Джерси: Прентис-Холл. 2008, с. 300. ISBN 0-13-613550-1
- ^ Хани (2009). «Исследование репликации» . Архивировано из оригинала 2 июня 2012 года . Проверено 27 октября 2011 г.
- ^ Меткалф, Нью-Хэмпшир (февраль 2011 г.). «Сэр Джеффри Маршалл (1887–1982): врач-респиратор, катализатор развития анестезии, врач премьер-министра и короля, а также командир баржи времен Первой мировой войны». Журнал медицинской биографии . 19 (1): 10–4. дои : 10.1258/jmb.2010.010019 . ПМИД 21350072 . S2CID 39878743 .
- ↑ Pharmabiz.com, 19 мая 2014 г., Мумбаи ISCR выпускает Руководство для участников клинических исследований в Международный день клинических испытаний. Архивировано 7 апреля 2023 г. в Wayback Machine (по состоянию на 20 мая 2014 г.).
- ^ Поттегорд, Антон; Хааструп, Майя Бруун; Сцена, Торе Бьеррегард; Хансен, Мортен Рикс; и др. (16 декабря 2014 г.). «Поиск юмористических и экстравагантных акронимов и совершенно неподходящих названий для важных клинических исследований (НАУЧНЫХ): качественное и количественное систематическое исследование» . БМЖ . 349 : g7092. дои : 10.1136/bmj.g7092 . ПМК 4267482 . ПМИД 25516539 .
- ↑ Перейти обратно: Перейти обратно: а б «Что такое клиническое исследование?» . Национальная медицинская библиотека, Национальные институты здравоохранения США. 1 марта 2019 г. Архивировано из оригинала 19 мая 2019 г. . Проверено 24 мая 2019 г.
- ^ Хелен С (2010). «Программы сострадательного использования ЕС (CUP): нормативно-правовая база и моменты, которые следует учитывать перед внедрением CUP» . Фарм Мед . 24 (4): 223–229. дои : 10.1007/BF03256820 . S2CID 31439802 .
- ^ Бреннан З. (5 июня 2013 г.). «CRO медленно переходят к адаптивному дизайну клинических исследований» . Аутсорсинг-pharma.com. Архивировано из оригинала 27 августа 2017 года . Проверено 5 января 2014 г.
- ^ «Адаптивные клинические испытания для преодоления исследовательских проблем» . Новости-medical.net. 17 сентября 2013 г. Архивировано из оригинала 11 августа 2020 г. . Проверено 4 января 2014 г.
- ^ Ван, Ширли С. (30 декабря 2013 г.). «Здоровье: ученые стремятся снизить стоимость и время испытаний лекарств - WSJ.com» . Онлайн.wsj.com. Архивировано из оригинала 14 марта 2016 года . Проверено 4 января 2014 г.
- ^ Хубер П.В. (12 ноября 2013 г.). Лекарство в кодексе: как закон 20-го века подрывает медицину 21-го века . Основные книги. ISBN 978-0-465-06981-1 .
- ^ Ланцет (июль 2009 г.). «Испытания фазы 0: платформа для разработки лекарств?» . Ланцет . 374 (9685): 176. doi : 10.1016/S0140-6736(09)61309-X . ПМИД 19616703 . S2CID 30939770 .
- ↑ Перейти обратно: Перейти обратно: а б «Клиническое исследование фазы IIa и фазы IIb» . www.musculardystrophyuk.org . Архивировано из оригинала 9 августа 2020 года . Проверено 10 августа 2020 г.
- ↑ Перейти обратно: Перейти обратно: а б с Ханнан Э.Л. (июнь 2008 г.). «Рандомизированные клинические испытания и наблюдательные исследования: рекомендации по оценке соответствующих сильных сторон и ограничений» . JACC. Сердечно-сосудистые вмешательства . 1 (3): 211–7. дои : 10.1016/j.jcin.2008.01.008 . ПМИД 19463302 .
- ^ Сесслер Д.И., Имри П.Б. (октябрь 2015 г.). «Методология клинических исследований 2: Наблюдательные клинические исследования» . Анестезия и анальгезия . 121 (4): 1043–51. doi : 10.1213/ANE.0000000000000861 . ПМИД 26378704 . S2CID 19333613 .
- ^ Элдридж С.М., Ланкастер Г.А., Кэмпбелл М.Дж., Табейн Л., Хоупвелл С., Коулман К.Л., Бонд К.М. (2016). «Определение технико-экономического обоснования и пилотных исследований при подготовке к рандомизированным контролируемым исследованиям: разработка концептуальной основы» . ПЛОС ОДИН . 11 (3): e0150205. Бибкод : 2016PLoSO..1150205E . дои : 10.1371/journal.pone.0150205 . ПМЦ 4792418 . ПМИД 26978655 .
- ^ «Активное управление/Активный компаратор» . 3 марта 2017 года. Архивировано из оригинала 8 апреля 2023 года . Проверено 31 октября 2017 г.
- ^ Янг, Сьюзен. «Foundation Medicine присоединяется к коалиции, стремящейся встряхнуть испытания лекарств от рака | Обзор технологий MIT» . Technologyreview.com. Архивировано из оригинала 13 ноября 2013 года . Проверено 14 ноября 2013 г.
- ^ «Официальный веб-сайт ICH» (PDF) . www.ich.org . Архивировано из оригинала (PDF) 21 сентября 2008 года.
- ^ «Официальный веб-сайт ICH: ICH» . ich.org . Архивировано из оригинала 25 мая 2011 года . Проверено 5 октября 2008 г.
- ↑ Перейти обратно: Перейти обратно: а б с «Узнайте о клинических исследованиях» . www.clinicaltrials.gov . Архивировано из оригинала 10 апреля 2012 года.
- ^ Мэлони, Деннис М. (1984). Защита объектов исследований на людях: Практическое руководство по федеральным законам и постановлениям . Бостон, Массачусетс: Springer US. п. 151. ИСБН 9781461327035 .
- ^ Дори, Фредерик (2011). «Кратко о статистике: статистическая мощь: что это такое и когда ее следует использовать?» . Клиническая ортопедия и связанные с ней исследования . 469 (2): 619–620. дои : 10.1007/s11999-010-1435-0 . ПМК 3018227 . ПМИД 20585913 .
- ^ Норвиц Э.Р., Гринберг Дж.А. (2011). «Одобрение FDA на использование лекарств во время беременности: тяжелая битва» . Обзоры по акушерству и гинекологии . 4 (2): 39–41. ПМК 3218552 . ПМИД 22102925 .
- ^ «Часто задаваемые вопросы | Онкологический центр Университета Аризоны» . Azcc.arizona.edu. Архивировано из оригинала 2 декабря 2013 года . Проверено 14 ноября 2013 г.
- ^ Мисета, Эд (17 декабря 2019 г.). «Янссен использует геозоны для мониторинга пациентов, участвующих в клинических исследованиях» . Клинический руководитель . Пенсильвания, США: VertMarkets. Архивировано из оригинала 27 января 2020 года . Проверено 26 января 2020 г. .
- ^ Унгер, Дж. М.; Кук, Э; Тай, Э; Блейер, А (2016). «Роль участия в клинических исследованиях рака: барьеры, доказательства и стратегии» . Учебная книга Американского общества клинической онкологии. Американское общество клинической онкологии. Ежегодное собрание . 35 (36). Американское общество клинической онкологии: 185–198. дои : 10.1200/EDBK_156686 . ПМЦ 5495113 . ПМИД 27249699 .
- ^ Вайс С.К., Роуэлл Р., Крохмаль Л. (2008). «Влияние сезонности на проведение клинических исследований в дерматологии». Клиники по дерматологии . 26 (5): 565–9. doi : 10.1016/j.clindermatol.2008.01.016 . ПМИД 18755376 .
- ^ Хан Ю, Тилли С. «Сезонность: логистические проблемы менеджера клинических исследований» (PDF) . Фарм-Олам Интернешнл. Архивировано из оригинала (PDF) 15 июля 2011 года . Проверено 26 апреля 2010 г.
- ^ Маршалл, Меган (19 декабря 2019 г.). «Внедрение QbD в ваше клиническое исследование? 4 вопроса, на которые нужно ответить в первую очередь» . Клинический руководитель . Пенсильвания: VertMarkets. Архивировано из оригинала 27 января 2020 года . Проверено 26 января 2020 г. .
- ^ Ян, ZJ и др. (2010). «Мотивация для поиска и обработки медицинской информации о наборе участников в клинические исследования». Health Communication 25(5): 423–436.
- ^ «BIO будет использовать ViS Analytics для оптимизации педиатрических клинических исследований - WSJ.com» . Онлайн.wsj.com. 7 мая 2013 года. Архивировано из оригинала 4 февраля 2014 года . Проверено 14 ноября 2013 г.
- ^ Группа стратегии в области медико-биологических наук, Синдицированная публикация «Использование технологий клинических испытаний, предпочтения при покупке и перспективы роста», май 2009 г.
- ^ «Результаты электронных отчетов пациентов (ePRO) – изменение облика клинических исследований» . Med-Quest.org . Архивировано из оригинала 21 мая 2015 года . Проверено 20 мая 2015 г.
- ↑ Перейти обратно: Перейти обратно: а б Эванс, Скотт Р. (2010). «Общие статистические проблемы в клинических исследованиях» . Журнал экспериментального инсульта и трансляционной медицины . 3 (1): 1–7. doi : 10.6030/1939-067x-3.1.1 (неактивен 17 марта 2024 г.). ISSN 1939-067X . ПМК 3059317 . ПМИД 21423790 .
{{cite journal}}: CS1 maint: DOI неактивен по состоянию на март 2024 г. ( ссылка ) - ↑ Перейти обратно: Перейти обратно: а б Эванс-младший, Швейцария; Ильдстад, ST, ред. (2001). «Статистические подходы к анализу небольших клинических исследований». Проблемы и проблемы малых клинических исследований; В: Статистические подходы к анализу небольших клинических исследований . Издательство национальных академий (США). Архивировано из оригинала 15 апреля 2023 года . Проверено 7 ноября 2022 г.
- ^ Гордон, Э.Дж.; Прохаска, ТР (2006). «Этика отказа от участия в исследовании» . Ответственность в исследованиях . 13 (4): 285–309. дои : 10.1080/08989620600848645 . ПМЦ 9527709 . ПМИД 17849641 .
- ^ «Законопроект № 2328» (PDF) . Архивировано из оригинала (PDF) 2 декабря 2007 года . Проверено 19 ноября 2007 г.
- ^ Хайден ЕС (ноябрь 2014 г.). «Этическая дилемма испытаний лекарств от Эболы» . Природа . 515 (7526): 177–8. Бибкод : 2014Natur.515..177C . дои : 10.1038/515177a . ПМИД 25391940 .
- ^ Паттинсон, Шон Д. (2012). «Экстренные исследования и интересы участников» (PDF) . Международное медицинское право . 12 (2): 121–141. дои : 10.1177/0968533212465615 . S2CID 71853867 . Архивировано (PDF) из оригинала 9 апреля 2023 года . Проверено 13 июля 2019 г.
- ^ Спан, Паула (3 марта 2020 г.). «Когда исследование препарата внезапно заканчивается, добровольцам приходится справляться с ситуацией» . Нью-Йорк Таймс . ISSN 0362-4331 . Архивировано из оригинала 7 апреля 2023 года . Проверено 23 апреля 2020 г.
- ^ Мойнихан, Р. (2003). «Кто платит за пиццу? Переосмысление отношений между врачами и фармацевтическими компаниями. Часть 2: Распутывание» . Британский медицинский журнал . 326 (7400): 1193–1196. дои : 10.1136/bmj.326.7400.1193 . ПМК 1126054 . ПМИД 12775622 .
- ^ «Информация Hogan & Hartson о регистрации фармацевтических исследований» (PDF) . 3 марта 2008 г. Архивировано из оригинала (PDF) 25 июня 2008 г. . Проверено 2 июня 2008 г.
- ^ «Рост количества опровержений научных журналов побуждает к реформам» . Нью-Йорк Таймс . 16 апреля 2012 года. Архивировано из оригинала 18 июня 2018 года . Проверено 22 мая 2018 г.
- ^ Вулли К.Л., Лью Р.А., Стреттон С., Эли Дж.А., Брамич Н.Дж., Киз Дж.Р., Монк Дж.А., Вулли М.Дж. (июнь 2011 г.). «Отсутствие участия писателей-медиков и фармацевтической промышленности в публикациях, отозванных за неправомерное поведение: систематическое, контролируемое ретроспективное исследование». Текущие медицинские исследования и мнения . 27 (6): 1175–82. дои : 10.1185/03007995.2011.573546 . ПМИД 21473670 . S2CID 2583905 .
- ^ Сокс ХК, Ренни Д. (август 2008 г.). «Посевные испытания: просто скажи «нет» » . Анналы внутренней медицины . 149 (4): 279–80. дои : 10.7326/0003-4819-149-4-200808190-00012 . ПМИД 18711161 .
- ^ «Процесс разработки и утверждения (лекарственные препараты)» . Управление по контролю за продуктами и лекарствами США. Архивировано из оригинала 10 июня 2018 года . Проверено 22 мая 2018 г.
- ^ «Информационный листок-руководство для IRB, клинических исследователей и спонсоров: проверки FDA клинических исследователей» (PDF) . Управление по контролю за продуктами и лекарствами . Июнь 2010 г. Архивировано (PDF) из оригинала 11 июня 2014 г. Проверено 16 октября 2014 г.
- ^ Давидофф Ф., ДеАнджелис С.Д., Дразен Дж.М., Николлс М.Г., Хоуи Дж., Хойгаард Л., Хортон Р., Котцин С., Найленна М., Овербеке А.Дж., Сокс ХК, Ван дер Вейден М.Б., Уилкс М.С. (сентябрь 2001 г.). «Спонсорство, авторство и ответственность» . CMAJ . 165 (6): 786–8. ПМК 81460 . ПМИД 11584570 . Архивировано из оригинала 15 апреля 2023 года . Проверено 27 февраля 2011 г.
- ^ Манси, Бернадетт А.; Кларк, Джули; Дэвид, Фрэнк С.; Гезелл, Томас М.; Глассер, Сьюзен; Гонсалес, Джон; Халлер, Дэниел Г.; Лейн, Кристина; Миллер, Чарльз Л.; Муни, ЛаВерн А.; Зечевич, Майя (май 2012 г.). «Десять рекомендаций по устранению разрыва в доверии к отчетности о клинических исследованиях, спонсируемых промышленностью: совместный взгляд журнала и фармацевтической промышленности» . Труды клиники Мэйо . 87 (5): 424–429. дои : 10.1016/j.mayocp.2012.02.009 . ISSN 1942-5546 . ПМЦ 3538468 . ПМИД 22560521 .
- ^ Тоэн, Маурисио (сентябрь 2013 г.). «Доверие к клиническим исследованиям, финансируемым промышленностью» . Международный журнал нейропсихофармакологии . 16 (8): 1879–1884. дои : 10.1017/S1461145713000461 . ISSN 1469-5111 . ПМИД 23672957 .
- ^ Тоэн, Маурисио (1 февраля 2017 г.). «Руководство по лечению биполярных расстройств и важность правильного дизайна клинических исследований» . Международный журнал нейропсихофармакологии . 20 (2): 95–97. дои : 10.1093/ijnp/pyx002 . ISSN 1469-5111 . ПМК 5356994 . ПМИД 28927197 .
- ^ Тоэн, Маурисио (2018). «Тяжесть симптомов в клинических рекомендациях по мании и последствия дизайна исследования» . Биполярные расстройства . 20 (2): 171–172. дои : 10.1111/bdi.12598 . ISSN 1399-5618 . ПМИД 29327798 . Архивировано из оригинала 8 апреля 2023 года . Проверено 27 мая 2021 г.
- ^ Тоэн, Маурисио (2013). «Доверие к клиническим исследованиям, финансируемым промышленностью» . Международный журнал нейропсихофармакологии . 16 (8): 1879–1884. дои : 10.1017/S1461145713000461 . ISSN 1469-5111 . ПМИД 23672957 .
- ↑ Перейти обратно: Перейти обратно: а б Эдвардс, Кэтрин М.; Кочхар, Сонали (2020). «Этика проведения клинических исследований в условиях вспышки» . Ежегодный обзор вирусологии . 7 (1): 475–494. doi : 10.1146/annurev-virology-013120-013123 . ПМИД 32212920 .
- ^ «Руководство NINDS по мониторингу в клинических исследованиях | Национальный институт неврологических расстройств и инсульта» . ninds.nih.gov . Архивировано из оригинала 9 января 2020 года . Проверено 25 ноября 2019 г.
- ^ «Руководство по обучению Совета по мониторингу данных и безопасности для исследований, инициированных следователями - Тафтс CTSI» . tuftsctsi.wpengine.com . 15 мая 2018 года. Архивировано из оригинала 7 апреля 2023 года . Проверено 25 ноября 2019 г.
- ^ Планирование и проведение клинических исследований – обзор . Международный журнал статистики и медицинской информатики. 2019. ISBN 9781096489085 .
- ^ «Для пациентов: информированное согласие на участие в клинических исследованиях» . Управление по контролю за продуктами и лекарствами США. 25 февраля 2016 года. Архивировано из оригинала 22 июля 2017 года . Проверено 9 августа 2017 г.
- ^ Руководство для промышленности: раскрытие финансовой информации клиническими исследователями , Управление по санитарному надзору за качеством пищевых продуктов и медикаментов , 20 марта 2001 г., заархивировано из оригинала 11 мая 2009 г. , получено 16 декабря 2019 г.
- ^ Ланг Т., Сирибаддана С. (2012). «Клинические испытания стали глобальными: хорошо ли это?» . ПЛОС Медицина . 9 (6): e1001228. дои : 10.1371/journal.pmed.1001228 . ПМЦ 3373653 . ПМИД 22719228 .
- ^ «Индия: главное место для неэтичных клинических испытаний» . Общие мечты . Архивировано из оригинала 15 декабря 2007 года . Проверено 16 декабря 2007 г.
- ^ «Агентство фармацевтических препаратов и медицинского оборудования Pmda.go.jp» (PDF) Архивировано из оригинала (PDF) 17 декабря 2008 г.
- ^ «Официальный веб-сайт ICH: ICH» . www.ich.org . Архивировано из оригинала 30 июня 2007 года.
- ^ «Управление информацией о безопасности клинических исследований: отчет Рабочей группы VI CIOMS» . Публикации ЦИОМС . 2005. Архивировано из оригинала 15 августа 2019 года . Проверено 11 ноября 2017 г.
- ^ «Заявка на новое исследовательское лекарство (IND) – Окончательное правило: Требования к отчетности о безопасности новых лекарств для человека и биологических продуктов, а также Требования к отчетности о безопасности для исследований биодоступности и биоэквивалентности на людях» . FDA.gov . Центр оценки и исследования лекарств. 29 сентября 2010 г. Архивировано из оригинала 23 апреля 2019 г. . Проверено 11 ноября 2017 г.
- ^ «Руководство для промышленности и следователей. Требования к отчетности по безопасности для исследований IND и BA/BE. Руководство по соблюдению требований для малых предприятий» (PDF) . Управление по контролю за продуктами и лекарствами =. Декабрь 2012 г. Архивировано (PDF) из оригинала 16 января 2019 г. . Проверено 11 ноября 2017 г.
- ^ «Оценка безопасности для руководства по отчетности о безопасности IND для промышленности» (PDF) . Управление по контролю за продуктами и лекарствами. Декабрь 2015 г. Архивировано (PDF) из оригинала 4 мая 2017 г. Проверено 11 ноября 2017 г.
- ^ «Синтез фактических данных и метаанализ: отчет рабочей группы X CIOMS» . Публикации ЦИОМС. 2016. Архивировано из оригинала 15 августа 2019 года . Проверено 11 ноября 2017 г.
- ^ Серткая, Айлин; Вонг, Хуэй-Синг; Джессап, Эмбер; Белече, Тринидад (2016). «Основные факторы затрат на фармацевтические клинические испытания в США» . Клинические испытания . 13 (2): 117–126. дои : 10.1177/1740774515625964 . ПМИД 26908540 . S2CID 24308679 .
- ^ «Налоговый кредит на расходы на тестирование лекарств от редких заболеваний или состояний» . Управление по контролю за продуктами и лекарствами . 17 апреля 2001 года. Архивировано из оригинала 6 апреля 2007 года . Проверено 27 марта 2007 г.
- ^ Пол Дж., Сейб Р., Прескотт Т. (март 2005 г.). «Интернет и клинические испытания: предыстория, онлайн-ресурсы, примеры и проблемы» . Журнал медицинских интернет-исследований . 7 (1): e5. дои : 10.2196/jmir.7.1.e5 . ПМК 1550630 . ПМИД 15829477 .
- ^ Станкель, Лиэнн; Грейди, Кристина (май 2011 г.). «Больше, чем деньги: обзор литературы, исследующей здоровую мотивацию волонтеров» . Современные клинические исследования . 32 (3): 342–352. дои : 10.1016/j.cct.2010.12.003 . ПМЦ 4943215 . ПМИД 21146635 .
- ^ Лю Джей Джей, Дэвидсон Э, Шейх А (2011). «Достижение этнического разнообразия при наборе в суд» . Фарм Мед . 25 (4): 215–222. дои : 10.1007/BF03256863 . S2CID 19557355 . Архивировано из оригинала 14 ноября 2011 года.
- ^ Макдональд А.М., Найт Р.С., Кэмпбелл М.К., Энтвистл В.А., Грант А.М., Кук Дж.А., Эльбурн Д.Р., Фрэнсис Д., Гарсия Дж., Робертс И., Сноудон С. (апрель 2006 г.). «Что влияет на набор участников в рандомизированные контролируемые исследования? Обзор исследований, финансируемых двумя финансирующими агентствами Великобритании» . Испытания . 7 :9. дои : 10.1186/1745-6215-7-9 . ПМЦ 1475627 . ПМИД 16603070 .
- ^ «Волонтерство в клинических исследованиях» . Бостон, Массачусетс: CenterWatch. 2016. Архивировано из оригинала 25 ноября 2016 года . Проверено 26 ноября 2016 г.
- ^ «Разнообразие и инклюзивность в клинических исследованиях» . НИМГД . Архивировано из оригинала 18 марта 2023 года . Проверено 9 декабря 2022 г.
- ^ Конкель, Линдси (2015). «Расовые и этнические различия в научных исследованиях: проблема создания более разнообразных когорт» . Перспективы гигиены окружающей среды . 123 (12): А297–А302. дои : 10.1289/ehp.123-A297 . ISSN 0091-6765 . ПМК 4670264 . ПМИД 26625444 .
- ^ «Почти половина чернокожих пациентов с метастатическим раком молочной железы не информированы о клинических испытаниях» . 6 августа 2022 года. Архивировано из оригинала 7 апреля 2023 года . Проверено 7 апреля 2023 г.
- ^ Питтман, Корин А.; Рура, Рауль; Прайс, Кэрри; Лин, Фрэнк Р.; Марроне, Николь; Ниман, Кэрри Л. (1 июля 2021 г.). «Расовая/этническая и половая репрезентация в американских клинических исследованиях по лечению потери слуха у взрослых: систематический обзор» . JAMA Отоларингология – хирургия головы и шеи . 147 (7): 656–662. дои : 10.1001/jamaoto.2021.0550 . ISSN 2168-6181 . ПМИД 33885733 . S2CID 233351877 .
- ^ «Как остановить отсутствие разнообразия, подрывающее данные клинических испытаний» . Файнэншл Таймс . 18 января 2019 года. Архивировано из оригинала 10 декабря 2022 года . Проверено 26 февраля 2019 г.
- ^ «Клинические исследования болезни Паркинсона» . Искатель проб Fox . Проверено 14 ноября 2013 г.
- ^ «Медицинская информация в Интернете» . Мланет.орг. Архивировано из оригинала 3 октября 2013 года . Проверено 14 ноября 2013 г.
- ^ Кляйн, Гарри; Мазор, Тали; Сигел, Итан; Труханов Павел; Овалье, Андреа; Веккио Фитц, Кэтрин Дель; Цвислер, Закари; Кумари, Прити; Ван дер Вин, Бернд; Марриотт, Эрик; Гензель, Джейсон; Ю, Джойс; Албайрак, Адем; Барри, Сьюзен; Келлер, Рэйчел Б.; МакКонейл, Лаура Э.; Линдеман, Нил; Джонсон, Брюс Э.; Роллинз, Барретт Дж.; До, Хан Т.; Бердсли, Брайан; Шапиро, Джеффри; Гектор-Барри, Сюзанна; Митот, Джон; Шолл, Линетт; Линдси, Джеймс; Хассетт, Майкл Дж.; Черами, Итан (6 октября 2022 г.). «MatchMiner: платформа с открытым исходным кодом для точной медицины рака» . npj Прецизионная онкология . 6 (1): 69. дои : 10.1038/s41698-022-00312-5 . ISSN 2397-768X . ПМЦ 9537311 . ПМИД 36202909 .
- ↑ Перейти обратно: Перейти обратно: а б Ян З.Дж., МакКомас К.А., Гей Г.К., Леонард Дж.П., Данненберг А.Дж., Диллон Х. (2012). «Сравнение принятия решений больными раком и населением в целом: мысли, эмоции или социальное влияние?». Журнал медицинских коммуникаций . 17 (4): 477–94. дои : 10.1080/10810730.2011.635774 . ПМИД 22376222 . S2CID 1344880 .
- ↑ Перейти обратно: Перейти обратно: а б «Что нужно знать о программах сопоставления клинических исследований рака» . Рак.Нет . 14 марта 2023 года. Архивировано из оригинала 28 сентября 2023 года . Проверено 26 декабря 2023 г.
- ↑ Перейти обратно: Перейти обратно: а б «Как искать клинические исследования» . ClinicalTrials.gov, Национальная медицинская библиотека США. 2023. Архивировано из оригинала 26 декабря 2023 года . Проверено 26 декабря 2023 г.
- ^ Фунцилас Э., Цимбериду А.М., Во Х.Х., Курцрок Р. (август 2022 г.). «Дизайн клинических исследований в эпоху точной медицины» . Геномная медицина . 14 (1): 101. дои : 10.1186/s13073-022-01102-1 . ПМЦ 9428375 . ПМИД 36045401 .
- ^ Флери МЭ (2023). «Консенсусные рекомендации по улучшению условий проведения клинических исследований рака» . Рак . 130 (1): 11–15. дои : 10.1002/cncr.35034 . ПМИД 37851508 .
- ^ Ван Норман, Джорджия (27 апреля 2021 г.). «Децентрализованные клинические исследования» . JACC: Основы трансляционной науки . 6 (4): 384–387. дои : 10.1016/j.jacbts.2021.01.011 . ПМК 8093545 . ПМИД 33997523 .
Внешние ссылки [ править ]
- Международная конференция по гармонизации технических требований к регистрации лекарственных средств для человеческого применения , руководство по регулированию клинических исследований
- ClinicalTrials.gov, всемирная база данных зарегистрированных клинических исследований ; Национальная медицинская библиотека США
- Кокрейновский центральный регистр контролируемых исследований (CENTRAL) ; концентрированный источник библиографических отчетов о рандомизированных контролируемых исследованиях
- ClinicalTrials.eu, Европейская информационная сеть по клиническим исследованиям ; Клинические испытания легко понять.