Биоинформатика


Биоинформатика ( / ˌ b aɪ . Oʊ ˌ ɪ n f ər ˈ m æ t ɪ k s / ) - это междисциплинарная область науки , которая разрабатывает методы и программные инструменты для понимания биологических данных, особенно когда наборы данных являются большими и сложными. Bioinformatics использует биологию , химию , физику , информатику , компьютерное программирование , информационную инженерию , математику и статистику для анализа и интерпретации биологических данных . Последующий процесс анализа и интерпретации данных часто называют вычислительной биологией , хотя различие между двумя терминами часто оспаривается.
Вычислительные, статистические и компьютерные методы программирования использовались для компьютерного моделирования анализа биологических запросов. Они включают повторный используемый специфический анализ «трубопроводы», особенно в области геномики , например, путем идентификации генов и отдельных нуклеотидных полиморфизмов ( SNP ). Эти трубопроводы используются для лучшего понимания генетической основы заболевания, уникальных адаптаций, желательных свойств (особенно у сельскохозяйственных видов) или различий между популяциями. Биоинформатика также включает протеомику , которая пытается понять организационные принципы в нуклеиновой кислоты и белков . последовательностях [ 1 ]
изображений и Обработка сигнала позволяет извлечь полезные результаты из больших объемов необработанных данных. В области генетики он помогает в секвенировании и аннотировании геномов и их наблюдаемых мутациях . Биоинформатика включает в себя добычу текста биологической литературы и развитие биологических и генных онтологий для организации и запроса биологических данных. Это также играет роль в анализе экспрессии и регуляции генов и белка. Инструменты биоинформатики помогают сравнивать, анализировать и интерпретировать генетические и геномные данные и, в целом, в понимании эволюционных аспектов молекулярной биологии. На более интегративном уровне он помогает анализировать и каталогизировать биологические пути и сети, которые являются важной частью системной биологии . В структурной биологии он помогает в моделировании и моделировании ДНК, [ 2 ] РНК, [ 2 ] [ 3 ] белки [ 4 ] а также биомолекулярные взаимодействия. [ 5 ] [ 6 ] [ 7 ] [ 8 ]
История
[ редактировать ]Первое определение термина биоинформатика было придумано Паулиеном Хогевегом и Беном Хеспером в 1970 году, чтобы ссылаться на изучение информационных процессов в биотических системах. [ 9 ] [ 10 ] [ 11 ] [ 12 ] [ 13 ] Это определение поместило биоинформатику как поле, параллельной биохимии (изучение химических процессов в биологических системах). [ 10 ]
Биоинформатика и вычислительная биология включали анализ биологических данных, особенно последовательностей ДНК, РНК и белка. В области биоинформатики наблюдалось взрывной рост, начиная с середины 1990-х годов, обусловленной в основном проектом генома человека и быстрыми успехами в технологии секвенирования ДНК. [ Цитация необходима ]
Анализ биологических данных для получения значимой информации включает в себя написание и запуск программных программ, которые используют алгоритмы из теории графов , искусственного интеллекта , мягких вычислений , интеллектуального анализа данных , обработки изображений и компьютерного моделирования . Алгоритмы, в свою очередь, зависят от теоретических оснований, таких как дискретная математика , теория управления , теория системы , теория информации и статистика . [ Цитация необходима ]
Последовательности
[ редактировать ]

С момента завершения проекта генома человека произошел огромный прогресс в сфере скорости и затрат, причем некоторые лаборатории способны последовать более 100 000 миллиардов баз в год, а полный геном может быть секвенирован за 1000 долларов или меньше. [ 14 ]
Компьютеры стали важными в молекулярной биологии, когда белковые последовательности стали доступны после того, как Фредерик Сэнгер определил последовательность инсулина в начале 1950 -х годов. [ 15 ] [ 16 ] Сравнение нескольких последовательностей вручную оказалось непрактичным. Маргарет Оукли Дейхофф , пионер в поле, [ 17 ] Скомпилировал одну из первых баз данных последовательностей белка, первоначально опубликованной как книги [ 18 ] а также методы выравнивания последовательностей и молекулярной эволюции . [ 19 ] Другим ранним участником биоинформатики был Элвин А. Кабат , который пионеровал анализ биологических последовательностей в 1970 году с его всесторонними объемами последовательностей антител, выпущенных в Интернете с Tai Te Wu между 1980 и 1991 годами. [ 20 ]
В 1970 -х годах новые методы секвенирования ДНК были применены к бактериофагам MS2 и ØX174, а затем расширенные нуклеотидные последовательности были проанализированы с помощью информационных и статистических алгоритмов. Эти исследования показали, что хорошо известные особенности, такие как сегменты кодирования и триплетный код, выявлены в простом статистическом анализе и были доказательством концепции того, что биоинформатика была бы проницательностью. [ 21 ] [ 22 ]
Цели
[ редактировать ]Чтобы изучить, как нормальная клеточная активность изменяется в разных болезненных состояниях, необработанные биологические данные должны быть объединены, чтобы сформировать всестороннюю картину этих действий. Поэтому [ когда? ] , область биоинформатики развивалась так, что наиболее насущная задача теперь включает анализ и интерпретацию различных типов данных. Это также включает в себя нуклеотидные и аминокислотные последовательности , белковые домены и белковые структуры . [ 23 ]
Важные субдисциплинары в биоинформатике и вычислительной биологии включают:
- Разработка и реализация компьютерных программ для эффективного доступа, управления и использования различных типов информации.
- Разработка новых математических алгоритмов и статистических мер для оценки отношений между членами крупных наборов данных. Например, существуют методы определения местонахождения гена в последовательности, для прогнозирования структуры и/или функции белка, а также для кластера белковых последовательностей в семейства родственных последовательностей.
Основной целью биоинформатики является повышение понимания биологических процессов. То, что отличает его от других подходов, - это сосредоточение на разработке и применении вычислительных интенсивных методов для достижения этой цели. Примеры включают в себя: распознавание шаблонов , интеллектуальный анализ данных , алгоритмы машинного обучения и визуализацию . Основные исследовательские усилия в этой области включают выравнивание последовательностей , обнаружение генов , сборку генома , дизайн лекарственного средства , обнаружение лекарственного средства , выравнивание структуры белка , прогнозирование структуры белка , прогнозирование экспрессии генов и взаимодействия белка-белка , исследования ассоциации по всему геному , моделирование эволюции и клеточное деление/митоз.
Биоинформатика влечет за собой создание и развитие баз данных, алгоритмов, вычислительных и статистических методов, а также теории для решения формальных и практических задач, возникающих в результате управления и анализа биологических данных.
За последние несколько десятилетий быстрое развитие технологий и разработок в области геномных и других молекулярных исследований в области информационных технологий объединилось для получения огромного объема информации, связанной с молекулярной биологией. Биоинформатика - это название, данное этим математическим и вычислительным подходам, используемым для получения понимания биологических процессов.
Общие действия в биоинформатике включают картирование и анализ последовательностей ДНК и белков, выравнивание последовательностей ДНК и белков для их сравнения, а также создание и просмотр трехмерных моделей белковых структур.
Анализ последовательности
[ редактировать ]С тех пор, как бактериофажный фаг φ-X174 был секвенирован в 1977 году, [ 24 ] Последовательности ДНК тысяч организмов были декодированы и хранятся в базах данных. Эта информация о последовательности анализируется для определения генов, которые кодируют белки , гены РНК, регуляторные последовательности, структурные мотивы и повторяющиеся последовательности. Сравнение генов внутри вида или между различными видами может показать сходство между функциями белка или отношениями между видами (использование молекулярной систематики для построения филогенетических деревьев ). С растущим объемом данных давно стало нецелесообразным анализировать последовательности ДНК вручную. Компьютерные программы , такие как взрыв, обычно используются для поиска последовательностей - за 2008 год, из более чем 260 000 организмов, содержащих более 190 миллиардов нуклеотидов . [ 25 ]
Секвенирование ДНК
[ редактировать ]Перед тем, как последовательности могут быть проанализированы, они получены из банка хранения данных, таких как GenBank. Секвенирование ДНК по-прежнему остается нетривиальной проблемой, так как необработанные данные могут быть шумными или затронуты слабыми сигналами. Алгоритмы были разработаны для базы, призывающих к различным экспериментальным подходам к секвенированию ДНК.
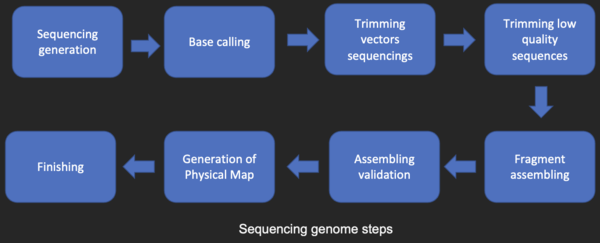
Сборка последовательности
[ редактировать ]Большинство методов секвенирования ДНК производят короткие фрагменты последовательности, которые необходимо собрать для получения полных последовательностей генов или генома. Метод секвенирования ружья (используемый Институтом геномных исследований (TIGR) для последовательности первого бактериального генома, Haemophilus influenzae ) [ 26 ] генерирует последовательности многих тысяч небольших фрагментов ДНК (в диапазоне от 35 до 900 нуклеотидов, в зависимости от технологии секвенирования). Концы этих фрагментов перекрываются и при правильном выровнении программой сборки генома могут использоваться для реконструкции полного генома. Секвенирование ружья быстро дает данные последовательности, но задача сборки фрагментов может быть довольно сложной для более крупных геномов. Для генома, столь же большого, как геном человека , для сборки фрагментов может потребоваться много дней времени процессора на многопроцессорных компьютерах, и полученная сборка обычно содержит многочисленные промежутки, которые должны быть заполнены позже. Секвенирование дробовика-это метод выбора практически для всех секвенированных геномов (а не методов спуска цепи или химического деградации), а алгоритмы сборки генома являются критической областью исследования биоинформатики.
Аннотация генома
[ редактировать ]В геномике . аннотация относится к процессу маркировки остановки и начала областей генов и других биологических особенностей в последовательности секвенированной ДНК Многие геномы слишком велики, чтобы быть аннотированными вручную. Поскольку скорость секвенирования превышает скорость аннотации генома, аннотация генома стала новым узким местом в биоинформатике [ когда? ] .
Аннотация генома может быть классифицирована на три уровня: уровни нуклеотида , белка и процесса.
Обнаружение генов является главным аспектом аннотации на уровне нуклеотидов. Для сложных геномов комбинация прогнозирования генов AB initio и сравнения последовательностей с экспрессированными базами данных последовательностей и других организмов может быть успешной. Аннотация на уровне нуклеотидов также позволяет интегрировать последовательность генома с другими генетическими и физическими картами генома.
Основная цель аннотации уровня белка состоит в том, чтобы назначить функцию белковых продуктов генома. Базы данных белковых последовательностей и функциональных доменов и мотивов используются для этого типа аннотации. Около половины прогнозируемых белков в новой последовательности генома, как правило, не имеют очевидной функции.
Понимание функции генов и их продуктов в контексте клеточной физиологии и организма является целью аннотации на уровне процесса. Препястью аннотации на уровне процесса было несоответствие терминов, используемых различными модельными системами. Консорциум генной онтологии помогает решить эту проблему. [ 27 ]
Первое описание комплексной системы аннотации было опубликовано в 1995 году [ 26 ] Институтом геномных исследований , который выполнял первое полное секвенирование и анализ генома свободноживущего (не симбиотического ) организма, бактерий гриппов Haemophilus . [ 26 ] Система идентифицирует гены, кодирующие все белки, переносят РНК, рибосомальные РНК, чтобы выполнить начальные функциональные назначения. Программа Genemark , обучающаяся найти кодирующие белки гены в гриппе Haemophilus, постоянно меняется и улучшается.
Следуя целям, которые проект генома человека оставил после его закрытия в 2003 году, проект ENCODE был разработан Национальным институтом исследований генома человека . Этот проект представляет собой совместный сбор данных функциональных элементов генома человека, в котором используются технологии ДНК-секвенирования ДНК следующего поколения и геномные массивы плитки, технологии, способные автоматически генерировать большие объемы данных при резко сниженной стоимости на базу, но с тем же Точность (ошибка базового вызова) и Fidelity (ошибка сборки).
Прогноз функции генов
[ редактировать ]В то время как аннотация генома в первую очередь основана на сходстве последовательностей (и, следовательно, гомологии ), другие свойства последовательностей могут использоваться для прогнозирования функции генов. Фактически, большинство методов прогнозирования функции генной функции сосредоточены на белковых последовательностях, поскольку они являются более информативными и более богатыми объектами. Например, распределение гидрофобных аминокислот предсказывает трансмембранные сегменты в белках. Однако прогноз функции белка также может использовать внешнюю информацию, такую как экспрессии данные генов (или белок), структуру белка или белковые взаимодействия . [ 28 ]
Вычислительная эволюционная биология
[ редактировать ]Эволюционная биология - это изучение происхождения и спуска видов , а также их изменения с течением времени. Информатика помогла эволюционным биологам, позволяя исследователям:
- проследить эволюцию большого числа организмов путем измерения изменений в их ДНК , а не только с помощью физической таксономии или физиологических наблюдений,
- Сравните целые геномы , которые позволяют изучать более сложные эволюционные события, такие как дупликация генов , горизонтальный перенос генов и прогнозирование факторов, важных для бактериального видообразования ,
- Создайте сложные модели генетики вычислительной популяции, чтобы предсказать результаты системы с течением времени [ 29 ]
- отслеживать и обмениваться информацией о все большем количестве видов и организмов
Будущая работа старается реконструировать ныне более сложное дерево жизни . [ Согласно кому? ]
Сравнительная геномика
[ редактировать ]Ядром сравнительного анализа генома является установление соответствия между генами ( ортологическим анализом) или другими геномными особенностями в разных организмах. Межгреномные карты создаются, чтобы проследить эволюционные процессы, ответственные за дивергенцию двух геномов. Множество эволюционных событий, действующих на различных организационных уровнях, формируют эволюцию генома. На самом низком уровне точечные мутации влияют на отдельные нуклеотиды. На более высоком уровне крупные хромосомные сегменты подвергаются дублированию, боковой переносе, инверсии, транспозиции, делеции и вставке. [ 30 ] Целые геномы участвуют в процессах гибридизации, полиплоидризации и эндосимбиоза , которые приводят к быстрому видообразованию. Сложность эволюции генома представляет много интересных проблем для разработчиков математических моделей и алгоритмов, которые обращаются к спектру алгоритмических, статистических и математических методов, начиная от точных эвристических , фиксированных параметров и алгоритмов приближения для проблем, основанных на моделях паризоны до Markatov . Алгоритмы сети Монте -Карло для байесовского анализа проблем, основанных на вероятностных моделях.
Многие из этих исследований основаны на обнаружении гомологии последовательности для назначения последовательностей семействам белков . [ 31 ]
Геномика Пан
[ редактировать ]Pan Genomics - это концепция, представленная в 2005 году Tettelin и Medini. Pan Genome - это полный генный репертуар конкретной монофилетической таксономической группы. Несмотря на то, что изначально применяется к тесно связанным штаммам вида, он может применяться к более широкому контексту, таким как род, тип и т. Д. Гены домашнего хозяйства, жизненно важные для выживания), а также невидимый/гибкий геном: набор генов, не присутствующих во всех изучаемых или некоторых геномах. BPGA BioInformatics Tool BPGA можно использовать для характеристики генома PAN бактериальных видов. [ 32 ]
Генетика болезней
[ редактировать ]По состоянию на 2013 год существование эффективной высокопроизводительной технологии секвенирования следующего поколения позволяет идентифицировать множество различных заболеваний человека. Простое наследство Менделяна наблюдалось при более чем 3000 расстройствах, которые были выявлены при онлайн -наследстве в базе данных MAN, но сложные заболевания сложнее. Исследования ассоциации обнаружили много отдельных генетических областей, которые индивидуально связаны с сложными заболеваниями (такими как бесплодие , [ 33 ] рак молочной железы [ 34 ] и болезнь Альцгеймера [ 35 ] ), а не единственная причина. [ 36 ] [ 37 ] В настоящее время существует много проблем с использованием генов для диагностики и лечения, например, как мы не знаем, какие гены важны, или насколько стабилен выбор, который обеспечивает алгоритм. [ 38 ]
Исследования ассоциаций по всему геному успешно выявили тысячи общих генетических вариантов для сложных заболеваний и признаков; Однако эти общие варианты объясняют только небольшую долю наследуемости. [ 39 ] Редкие варианты могут объяснить некоторую пропавшую наследуемость . [ 40 ] Крупномасштабные исследования секвенирования цельного генома быстро секвенировали миллионы цельных геномов, и в таких исследованиях выявили сотни миллионов редких вариантов . [ 41 ] Функциональные аннотации предсказывают эффект или функцию генетического варианта и помогают определить приоритеты редких функциональных вариантов, а включение этих аннотаций может эффективно повысить силу генетической ассоциации анализа редких вариантов исследований секвенирования цельного генома. [ 42 ] Были разработаны некоторые инструменты для обеспечения анализа редких вариантов ассоциации в одном для данных для данных секвенирования всего генома, включая интеграцию данных генотипа и их функциональные аннотации, анализ ассоциации, краткое изложение результатов и визуализация. [ 43 ] [ 44 ] Метаанализ исследований секвенирования цельного генома обеспечивает привлекательное решение проблемы сбора больших размеров выборки для обнаружения редких вариантов, связанных со сложными фенотипами. [ 45 ]
Анализ мутаций при раке
[ редактировать ]При раке геномы пораженных клеток переставляются сложными или непредсказуемыми способами. В дополнение к однонуклеотидным полиморфизму массивы, идентифицирующие точечные мутации , которые вызывают рак, олигонуклеотидные микрочипы могут использоваться для идентификации хромосомных усилений и потерь (называемый сравнительной геномной гибридизацией ). Эти методы обнаружения генерируют терабайты данных на эксперимент. Часто обнаруживается, что данные содержат значительную вариабельность или шум , и, таким образом, разработаны скрытые модели Маркова и методы анализа точки изменения для вывода реальных изменений числа копий . [ Цитация необходима ]
Два важных принципа могут быть использованы для идентификации рака путем мутаций в экзоме . Во -первых, рак - это заболевание накопленных соматических мутаций в генах. Во -вторых, рак содержит мутации водителя, которые необходимо отличить от пассажиров. [ 46 ]
Дальнейшие улучшения в биоинформатике могут позволить классифицировать типы рака путем анализа мутаций, вызванных раком, в геноме. Кроме того, отслеживание пациентов, в то время как прогрессирование заболевания может быть возможным в будущем с последовательности образцов рака. Другим типом данных, которые требуют новой разработки информатики, является анализ поражений , которые, как оказались, являются повторяющимися среди многих опухолей. [ 47 ]
Экспрессия гена и белка
[ редактировать ]Анализ экспрессии генов
[ редактировать ]Экспрессия RNA-Seq,, Seq ,, многих генов можно определить путем измерения уровней мРНК с помощью множественных методов, включая микрочипы , экспрессированное секвенирование последовательности кДНК (EST), последовательный анализ секвенирования TAG экспрессии генов (SAGE), массово параллельное секвенирование сигнатуры (MPSS), Seq Seq,, Seq,, Seq,, Seq, Seq, Seq, Seq, Seq, Seq, Seq, Seq, Seq, Seq, Seq, Seq, Seq, Seq, Seq, Seq, Seq, Seq, Seq, Seq, Seq, Seq, Seq , Также известный как «секвенирование с дробовиком всего транскрипта» (WTSS) или различные приложения мультиплексированной гибридизации in-situ. Все эти методы чрезвычайно подвержены шуму и/или подвержены смещению в биологическом измерении, а основная область исследований в вычислительной биологии включает в себя разработку статистических инструментов для отделения сигнала от шума в исследованиях экспрессии генов с высокой пропускной способностью. [ 48 ] Такие исследования часто используются для определения генов, участвующих в расстройстве: можно сравнить данные микрочипов из раковых эпителиальных клеток с данными из незвуковых клеток, чтобы определить транскрипты, которые активируются и понижаются в определенной популяции раковых клеток Полем

Анализ экспрессии белка
[ редактировать ]Белковые микрочипы с высокой пропускной способностью (HT) и масс -спектрометрия (MS) могут обеспечить снимок белков, присутствующих в биологическом образце. Первый подход сталкивается с аналогичными проблемами, что и с микрочипами, нацеленными на мРНК, последний включает в себя проблему сопоставления больших объемов массовых данных с прогнозируемыми массами из баз данных последовательностей белков, а также сложный статистический анализ образцов, когда обнаруживаются множественные неполные пептиды из каждого белка. Локализация клеточного белка в тканевом контексте может быть достигнута сроком с аффинной протеомикой, отображаемой в виде пространственных данных, основанных на иммуногистохимии и микрочипах ткани . [ 49 ]
Анализ регулирования
[ редактировать ]Регуляция генов - это сложный процесс, в котором сигнал, такой как внеклеточный сигнал, такой как гормон , в конечном итоге приводит к увеличению или снижению активности одного или нескольких белков . Методы биоинформатики были применены для изучения различных этапов в этом процессе.
Например, экспрессия генов может регулироваться близлежащими элементами в геноме. Промоторный анализ включает в себя идентификацию и изучение мотивов последовательности в ДНК, окружающей область кодирования белка гена. Эти мотивы влияют на степень, в которой эта область транскрибируется в мРНК. Элементы энхансеры , далеко от промотора, также могут регулировать экспрессию генов посредством трехмерного цикла взаимодействия. Эти взаимодействия могут быть определены с помощью биоинформатического анализа экспериментов по захвату конформации хромосом .
Данные экспрессии могут быть использованы для определения регуляции генов: можно сравнить данные микрочипа с широким спектром состояний организма, чтобы формировать гипотезы о генах, участвующих в каждом состоянии. В одноклеточном организме можно сравнить стадии клеточного цикла , а также различные условия напряжения (тепловой шок, голода и т. Д.). Алгоритмы кластеризации могут затем применяться к данным экспрессии, чтобы определить, какие гены совместно экспрессируются. Например, вверх по течению регионов (промоторов) коэкспрессированных генов можно искать чрезмерные регуляторные элементы . Примерами кластеризационных алгоритмов, применяемых в кластеризации генов, являются K-средние кластеризация , самоорганизационные карты (SOMS), иерархическая кластеризация и методы кластеризации консенсуса .
Анализ клеточной организации
[ редактировать ]Было разработано несколько подходов для анализа местоположения органеллов, генов, белков и других компонентов в клетках. Категория генной онтологии , клеточная компонент , была разработана для захвата субклеточной локализации во многих биологических базах данных .
Микроскопия и анализ изображений
[ редактировать ]Микроскопические картинки позволяют определять местонахождение органеллов , а также молекулы, которые могут быть источником аномалий при заболеваниях.
Локализация белка
[ редактировать ]Поиск местоположения белков позволяет нам предсказать, что они делают. Это называется прогнозом функции белка . Например, если белок обнаружен в ядре , он может быть вовлечен в регуляцию генов или сплайсинг . Напротив, если белок обнаружен в митохондриях , он может быть вовлечен в дыхание или другие метаболические процессы . Доступны хорошо разработанные ресурсы прогнозирования субклеточной локализации белков , включая базы данных субклеточного местоположения белка и инструменты прогнозирования. [ 50 ] [ 51 ]
Ядерная организация хроматина
[ редактировать ]Данные из высокопроизводительных экспериментов по конформации хромосом , таких как HI-C (эксперимент) и Chia-Pet предоставить информацию о трехмерной структуре и ядерной организации хроматина , могут . Биоинформатические проблемы в этой области включают разделение генома на домены, такие как топологически ассоциирующие домены (TAD), которые организованы вместе в трехмерном пространстве. [ 52 ]
Структурная биоинформатика
[ редактировать ]
Поиск структуры белков является важным применением биоинформатики. Критическая оценка прогнозирования структуры белков (CASP) - это открытая конкуренция, где всемирные исследовательские группы представляют модели белков для оценки неизвестных моделей белка. [ 53 ] [ 54 ]
Аминокислотная последовательность
[ редактировать ]Линейная аминокислотная последовательность белка называется первичной структурой . Первичная структура может быть легко определена из последовательности кодонов на гене ДНК, которая кодирует для нее. У большинства белков первичная структура уникально определяет 3-мерную структуру белка в его нативной среде. Исключением является неправильно свернутый белок, участвующий в губчатой энцефалопатии крупного рогатого скота . Эта структура связана с функцией белка. Дополнительная структурная информация включает в себя вторичную , третичную и четвертичную структуру. Жизнеспособное общее решение для прогнозирования функции белка остается открытой проблемой. Большая часть усилий была направлена на эвристику, которая работает большую часть времени. [ Цитация необходима ]
Гомология
[ редактировать ]В геномной ветви биоинформатики гомология используется для прогнозирования функции гена: если последовательность гена A , чья функция известна, гомологична последовательности гена B, чья функция неизвестна, можно сделать вывод, что B может Поделиться функцией А. В структурной биоинформатике гомология используется для определения того, какие части белка важны в формировании структуры и взаимодействии с другими белками. Моделирование гомологии используется для прогнозирования структуры неизвестного белка из существующих гомологичных белков.
Одним из примеров этого является гемоглобин у людей и гемоглобин у бобовых ( лехемоглобин ), которые являются отдаленными родственниками из одной и той же белковой суперсемейства . Оба служат одной и той же целью транспортировки кислорода в организме. Хотя оба этих белка имеют совершенно разные аминокислотные последовательности, их белковые структуры практически идентичны, что отражает их почти идентичные цели и общего предка. [ 55 ]
Другие методы прогнозирования структуры белка включают белковое нити и De novo физическое моделирование (с нуля).
Другой аспект структурной биоинформатики включает использование белковых структур для виртуальных моделей скрининга, таких как модели количественной структур-активности и протеохемометрические модели (PCM). Кроме того, кристаллическая структура белка может использоваться при моделировании, например, лиганд-связывающих исследованиях и в исследованиях мутагенеза кремнеза.
в 2021 году глубокого обучения Программное обеспечение на основе алгоритмов на основе под названием Alphafold , разработанное Google DeepMind , значительно превосходит все другие методы программного обеспечения для прогнозирования [ 56 ] [ как? ] , и выпустил прогнозируемые структуры для сотен миллионов белков в базе данных структуры алфалолда. [ 57 ]
Биология сети и систем
[ редактировать ]Анализ сети стремится понять отношения в биологических сетях, таких как метаболические или белковые сети взаимодействия . Хотя биологические сети могут быть построены из одного типа молекулы или сущности (таких как гены), биология сети часто пытается интегрировать множество различных типов данных, таких как белки, мелкие молекулы, данные экспрессии генов и другие, которые все физически связаны , функционально или оба.
Системная биология включает в себя использование компьютерного моделирования клеточных , подсистем (таких как сети метаболитов и ферментов которые составляют метаболизм , передачи сигнала пути и регуляторные сети генов ) для анализа и визуализации сложных соединений этих клеточных процессов. Искусственная жизнь или виртуальная эволюция пытается понять эволюционные процессы посредством компьютерного моделирования простых (искусственных) форм жизни.
Молекулярные сети взаимодействия
[ редактировать ]
Десятки тысяч трехмерных белковых структур были определены с помощью рентгеновской кристаллографии и белковой ядерной магнитно-резонансной спектроскопии (белковой ЯМР) и центральным вопросом в структурной биоинформатике заключается 3D -формы, не выполняя по взаимодействию белка -белок эксперименты . Было разработано различные методы для решения проблемы стыковки белка и белка , хотя, похоже, в этой области еще предстоит проделать много работы.
Другие взаимодействия, встречающиеся в полевых условиях, включают белок -лиганд (включая препарат) и белок -пептид . Молекулярное динамическое моделирование движения атомов в отношении вращающихся связей является фундаментальным принципом вычислительных алгоритмов , называемых алгоритмами стыковки, для изучения молекулярных взаимодействий .
Информатика биоразнообразия
[ редактировать ]Информатика в области биоразнообразия посвящена сбору и анализу данных о биоразнообразии , таких как таксономические базы данных или микробиома данные . Примеры таких анализов включают филогенетику , нишевое моделирование , картирование видов , штрих -кодирование ДНК или видов инструменты идентификации . Растущая область также является макроэкологией , то есть изучение того, как биоразнообразие связано с экологией и воздействием человека, таким как изменение климата .
Другие
[ редактировать ]Анализ литературы
[ редактировать ]Огромное количество опубликованной литературы делает практически невозможным для людей, чтобы читать каждую статью, что приводит к разрозненным суб-полям исследований. Анализ литературы направлен на использование вычислительной и статистической лингвистики для добычи этой растущей библиотеки текстовых ресурсов. Например:
- Признание аббревиатуры-определить длинную форму и аббревиатуру биологических терминов
- Признание по имени-энту -признание биологических терминов, таких как названия генов
- Взаимодействие белка -белка - определить, какие белки взаимодействуют с какими белками из текста
Область исследований вытекает из статистики и вычислительной лингвистики .
Высокопроизводительный анализ изображений
[ редактировать ]с высоким содержанием информаций Вычислительные технологии используются для автоматизации обработки, количественной оценки и анализа больших количеств биомедицинских изображений . Современные системы анализа изображений наблюдателя могут повысить точность , объективность или скорость . Анализ изображений важен как для диагностики , так и для исследований. Некоторые примеры:
- Высокопроизводительная и высокая точности количественная оценка и субклеточная локализация ( скрининг с высоким содержанием , цитогистопатология, информатика биоимноя )
- Морфометрия
- Анализ и визуализация клинического изображения
- Определение схем воздушного потока в реальном времени в дыхательных легких живых животных
- Количественная оценка размера окклюзии в образах в реальном времени от развития и восстановления во время артериальных травм
- Создание поведенческих наблюдений из расширенных видеозаписей лабораторных животных
- инфракрасные измерения для определения метаболической активности
- Вывод клонов перекрывается в картировании ДНК , например, Sulston Score
Высокопроизводительный анализ данных отдельных ячеек
[ редактировать ]Вычислительные методы используются для анализа высокопроизводительных данных с низким измерением, например, полученными с помощью проточной цитометрии . Эти методы обычно включают поиск популяций клеток, которые имеют отношение к конкретному состоянию заболевания или экспериментальному состоянию.
Онтологии и интеграция данных
[ редактировать ]Биологические онтологии являются направленными ациклическими графами словари контролируемых . Они создают категории для биологических концепций и описаний, поэтому их можно легко проанализировать с помощью компьютеров. При категории категории можно получить добавленную стоимость из целостного и интегрированного анализа. [ Цитация необходима ]
Форективы OBO были попыткой стандартизировать определенные онтологии. Одной из наиболее распространенных является генная онтология , которая описывает функцию генов. Есть также онтологии, которые описывают фенотипы.
Базы данных
[ редактировать ]Базы данных важны для исследований и приложений биоинформатики. Базы данных существуют для многих различных типов информации, включая последовательности ДНК и белков, молекулярные структуры, фенотипы и биоразнообразие. Базы данных могут содержать как эмпирические данные (полученные непосредственно из экспериментов), так и прогнозируемые данные (полученные из анализа существующих данных). Они могут быть специфичными для конкретного организма, пути или молекулы, представляющей интерес. В качестве альтернативы они могут включать данные, собранные из нескольких других баз данных. Базы данных могут иметь разные форматы, механизмы доступа и быть государственными или частными.
Некоторые из наиболее часто используемых баз данных перечислены ниже:
- Используется в анализе биологических последовательностей: GenBank , Uniprot
- Используется в структурном анализе: банк данных белка (PDB)
- Используется при поиске семейств белков и поиска мотива : InterPro , PFAM
- Используется для последовательности следующего поколения: архив чтения последовательности
- Используется в сетевом анализе: базы данных метаболических путей ( KEGG , BIOCYC ), базы данных анализа взаимодействия, функциональные сети
- Используется в дизайне синтетических генетических цепей: Genocad [ Цитация необходима ]
Программное обеспечение и инструменты
[ редактировать ]Программные инструменты для биоинформатики включают простые инструменты командной строки, более сложные графические программы и автономные веб-сервисы. Они сделаны компаниями биоинформатики или государственными учреждениями.
Программное обеспечение для биоинформатики с открытым исходным кодом
[ редактировать ]Многие бесплатные программные инструменты и программные инструменты с открытым исходным кодом существуют и продолжали расти с 1980-х годов. [ 59 ] Сочетание постоянной потребности в новых алгоритмах для анализа новых типов биологических счетов, потенциала для инновационных экспериментов в Silico и свободно доступных баз открытого кодекса создал возможности для исследовательских групп для внесения вклад в обе биоинформатики независимо от финансирования . Инструменты с открытым исходным кодом часто выступают в качестве инкубаторов идей или плагинов, поддерживаемых сообществом в коммерческих приложениях. Они также могут обеспечить фактические стандарты и модели общих объектов для оказания помощи в проблеме интеграции биоинформации.
Программное обеспечение для биоинформатики с открытым исходным кодом включает в себя биоконкуртирующие , биоперные , биопитон , биоджавы , биодж , биоруби , биоклипс , эмбуп , .NET Bio, Orange с надстройкой биоинформатики, таверной Apache , Угене и Genocad .
Некоммерческий фонд открытой биоинформатики [ 59 ] и ежегодная конференция с открытым исходным кодом с открытым исходным кодом продвигает программное обеспечение для биоинформатики с открытым исходным кодом. [ 60 ]
Веб -сервисы в биоинформатике
[ редактировать ]Были разработаны интерфейсы на основе SOAP и покоя , чтобы позволить клиентским компьютерам использовать алгоритмы, данные и вычислительные ресурсы с серверов в других частях мира. Основное преимущество заключается в том, что конечным пользователям не нужно иметь дело с накладными расходами на обслуживание программного обеспечения и базы данных.
Основные услуги биоинформатики классифицируются по EBI на три категории: SSS (сервисы поиска последовательностей), MSA (выравнивание множественных последовательностей) и BSA (анализ биологических последовательности). [ 61 ] Доступность этих ориентированных на сервис биоинформатических ресурсов демонстрирует применимость веб-биоинформатических решений и варьируется от набора автономных инструментов с общим форматом данных под одним веб-интерфейсом, до интегративных, распределенных и расширяемых систем управления рабочими потоками биоинформатики. Полем
Системы управления рабочими процессами биоинформатики
[ редактировать ]Система управления рабочими процессами биоинформатики представляет собой специализированную форму системы управления рабочими процессами, разработанной специально для составления и выполнения серии шагов вычислительных или манипулирования данными или рабочего процесса в приложении биоинформатики. Такие системы предназначены для
- Предоставьте простую в использовании среду для самих ученых-приложений для создания собственных рабочих процессов,
- Предоставьте интерактивные инструменты для ученых, позволяющих им выполнять свои рабочие процессы и просматривать свои результаты в режиме реального времени,
- упростить процесс обмена и повторного использования рабочих процессов между учеными и
- Позвольте ученым отслеживать происхождение результатов выполнения рабочего процесса и шагов создания рабочих процессов.
Некоторые из платформ, предоставляемых этой услугой: Галактика , Кеплер , Таверна , Угене , Андурил , Улей .
Биокомпюрунные и биокомпингические объекты
[ редактировать ]В 2014 году Управление по санитарному надзору за продуктами и лекарствами США выступило на конференции, состоявшейся в кампусе Национального института здравоохранения Bethesda для обсуждения воспроизводимости в биоинформатике. [ 62 ] В течение следующих трех лет консорциум заинтересованных сторон регулярно встречался, чтобы обсудить, что станет биокомпьютевой парадигмой. [ 63 ] Эти заинтересованные стороны включали представителей правительства, промышленности и академических организаций. Лидеры сессий представляли многочисленные филиалы институтов и центров FDA и NIH, некоммерческие организации, включая проект Variome Human и Европейскую федерацию медицинской информатики , и исследовательские институты, включая Стэнфорд , Центр генома Нью-Йорка и Университет Джорджа Вашингтона .
Было решено, что биологическая парадигма будет в форме цифровых «лабораторных ноутбуков», которые позволяют воспроизводить, репликацию, обзор и повторное использование протоколов биоинформатики. Это было предложено, чтобы обеспечить большую непрерывность в исследовательской группе в ходе обычного потока персонала, одновременно продвигая обмен идей между группами. FDA США финансировало эту работу так, чтобы информация о трубопроводах была более прозрачной и доступной для их регулирующего персонала. [ 64 ]
В 2016 году группа переработала в NIH в Бетесде и обсудила потенциал для биокомпьютерного объекта , экземпляра биокомпьютевой парадигмы. Эта работа была скопирована как документ «Стандартный пробный применение», так и документ с предварительной обработкой, загруженную в Biorxiv. Биологентный объект позволяет разделить записи JSON, которые будут использоваться среди сотрудников, сотрудников и регулирующих органов. [ 65 ] [ 66 ]
Образовательные платформы
[ редактировать ]Биоинформатика преподается не только как личная степень магистра во многих университетах. Вычислительный характер биоинформатики придает его компьютерному и онлайн-обучению . [ 67 ] [ 68 ] Программные платформы, предназначенные для обучения концепциям и методам биоинформатики, включают в себя курсы Rosalind и онлайн, предлагаемые через биоинформатики Швейцарский институт учебного портала . Канадские семинары по биоинформатике предоставляют видео и слайды из учебных семинаров на своем веб -сайте по лицензии Creative Commons . Проект 4273π или проект 4273PI [ 69 ] Также предлагает образовательные материалы с открытым исходным кодом бесплатно. Курс проходит на недорогих компьютерах Raspberry Pi и использовался для обучения взрослых и школьных учеников. [ 70 ] [ 71 ] 4273 активно разрабатывается консорциумом ученых и исследовательских сотрудников, которые управляют биоинформатикой на уровне исследований с использованием компьютеров Raspberry PI и операционной системой 4273. [ 72 ] [ 73 ]
Платформы MOOC также предоставляют онлайн -сертификаты по биоинформатике и связанных с ними дисциплинам, включая Coursera специализацию биоинформатики в Калифорнийском университете, Сан -Диего , специализацию по науке о геномных данных в Университете Джона Хопкинса и анализ данных EDX для Life Sciences Xseries в Гарвардском университете .
Конференции
[ редактировать ]Есть несколько крупных конференций, которые связаны с биоинформатикой. Некоторые из наиболее заметных примеров являются интеллектуальные системы для молекулярной биологии (ISMB), Европейская конференция по вычислительной биологии (ECCB) и исследования в области вычислительной молекулярной биологии (рекомбийт).
Смотрите также
[ редактировать ]- Информатика биоразнообразия
- Биоинформатические компании
- Вычислительная биология
- Вычислительная биомоделирование
- Вычислительная геномика
- Кибербисезопасность
- Функциональная геномика
- База данных по болезням генов
- Медицинская информатика
- Международное общество вычислительной биологии
- Библиотека прыжков
- Список учреждений биоинформатики
- Список программного обеспечения для биоинформатики с открытым исходным кодом
- Список журналов биоинформатики
- Метаболомика
- Митомап
- Последовательность нуклеиновой кислоты
- Филогенетика
- Протеомика
Ссылки
[ редактировать ]- ^ Lesk Am (26 июля 2013 г.). «Биоинформатика» . Энциклопедия Британская . Архивировано из оригинала 14 апреля 2021 года . Получено 17 апреля 2017 года .
- ^ Jump up to: а беременный Sim AY, Minary P, Levitt M (июнь 2012 г.). «Моделирование нуклеиновых кислот» . Современное мнение в структурной биологии . 22 (3): 273–8. doi : 10.1016/j.sbi.2012.03.012 . PMC 4028509 . PMID 22538125 .
- ^ Доусон WK, Maciejczyk M, Jankowska EJ, Bujnicki JM (июль 2016 г.). «Крупное моделирование РНК 3D-структуры» . Методы 103 : 138–56. Doi : 10.1016/j.ymeth.2016.04.026 . PMID 27125734 .
- ^ Kmiecik S, Gront D, Kolinski M, Wieteska L, Dawid AE, Kolinski A (июль 2016 г.). «Грубые модели белка и их применение» . Химические обзоры . 116 (14): 7898–936. Doi : 10.1021/acs.chemrev.6b00163 . PMID 2733362 .
- ^ Wong KC (2016). Вычислительная биология и биоинформатика: регуляция генов . CRC Press/Taylor & Francis Group. ISBN 978-1-4987-2497-5 .
- ^ Джойс А.П., Чжан С., Брэдли П., Хавранек Дж.Дж. (январь 2015). «Структурное моделирование белка: специфичность ДНК» . Брифинги в функциональной геномике . 14 (1): 39–49. doi : 10.1093/bfgp/elu044 . PMC 4366589 . PMID 25414269 .
- ^ Spiga E, Degiacomi MT, Dal Peraro M (2014). «Новые стратегии интегративного динамического моделирования макромолекулярной сборки». В Карабенчева-Кристовой Т (ред.). Биомолекулярное моделирование и моделирование . Достижения в области химии белка и структурной биологии. Тол. 96. Академическая пресса. С. 77–111. doi : 10.1016/bs.apcsb.2014.06.008 . ISBN 978-0-12-800013-7 Полем PMID 25443955 .
- ^ Ciemny M, Kurcinski M, Kamel K, Kolinski A, Alam N, Schueler-Furman O, et al. (Август 2018). «Белко-пептидная стыковка: возможности и проблемы» . Drug Discovery сегодня . 23 (8): 1530–1537. doi : 10.1016/j.drudis.2018.05.006 . PMID 29733895 .
- ^ Ouzounis CA, Valencia A (2003). «Ранняя биоинформатика: рождение дисциплины - личный взгляд» . Биоинформатика . 19 (17): 2176–2190. doi : 10.1093/bioinformatics/btg309 . PMID 14630646 .
- ^ Jump up to: а беременный Hogeweg P (2011). «Корни биоинформатики в теоретической биологии» . PLOS Computational Biology . 7 (3): E1002021. BIBCODE : 2011PLSCB ... 7E2021H . doi : 10.1371/journal.pcbi.1002021 . PMC 3068925 . PMID 21483479 .
- ^ Hesper B, Hogeweg P (1970). «Биоинформатика: концепция работы» [Биоинформатика: рабочая концепция]. Камелеон (на голландском языке). 1 (6): 28–29.
- ^ Hesper B, Hogeweg P (2021). «Биоинформатика: рабочая концепция. Перевод« биоинформатики: концепция работы »Б. Хеспера и П. Хогевега». Arxiv : 2111.11832v1 [ q-bio.ot ].
- ^ Hogeweg P (1978). «Моделирование роста клеточных форм». Симуляция . 31 (3): 90–96. doi : 10.1177/003754977803100305 . S2CID 61206099 .
- ^ Колби Б (2022). «Стоимость секвенирования целого генома» . Sequencing.com . Архивировано из оригинала 15 марта 2022 года . Получено 8 апреля 2022 года .
- ^ Сангер Ф., Таппи Х (1951). «Аминокислотная последовательность в фенилаланильной цепи инсулина. I. Идентификация нижних пептидов из частичных гидролизатов» . Биохимический журнал . 49 (4): 463–81. doi : 10.1042/bj0490463 . PMC 1197535 . PMID 14886310 .
- ^ Сангер Ф., Томпсон Эо (1953). «Аминокислотная последовательность в глицильной цепи инсулина. I. Идентификация нижних пептидов из частичных гидролизатов» . Биохимический журнал . 53 (3): 353–66. doi : 10.1042/bj0530353 . PMC 1198157 . PMID 13032078 .
- ^ Moody G (2004). Цифровой кодекс жизни: как биоинформатика революционизирует науку, медицину и бизнес . Хобокен, Нью -Джерси, США: Джон Вили и сыновья. ISBN 978-0-471-32788-2 .
- ^ Dayhoff MO, Eck RV, Chang Ma, Sochard MR (1965). Атлас белковой последовательности и структуры (PDF) . Серебряная весна, доктор медицинских наук, США: Национальный фонд биомедицинских исследований. LCCN 65-29342 .
- ^ Эк Р.В., Дейхофф Мо (апрель 1966 г.). «Эволюция структуры ферредоксина на основе живых реликвий примитивных аминокислотных последовательностей». Наука . 152 (3720): 363–6. Bibcode : 1966sci ... 152..363e . doi : 10.1126/science.152.3720.363 . PMID 17775169 . S2CID 23208558 .
- ^ Johnson G, Wu TT (январь 2000 г.). «База данных Kabat и ее приложения: через 30 лет после первого графика изменчивости» . Исследование нуклеиновых кислот . 28 (1): 214–8. doi : 10.1093/nar/28.1.214 . PMC 102431 . PMID 10592229 .
- ^ Эриксон Дж.В., Альтман Г.Г. (1979). «Поиск паттернов в нуклеотидной последовательности генома MS2». Журнал математической биологии . 7 (3): 219–230. doi : 10.1007/bf00275725 . S2CID 85199492 .
- ^ Шульман М.Дж., Стейнберг С.М., Уэстморленд Н. (февраль 1981 г.). «Функция кодирования нуклеотидных последовательностей может быть замечена статистическим анализом». Журнал теоретической биологии . 88 (3): 409–20. Bibcode : 1981jthbi..88..409s . doi : 10.1016/0022-5193 (81) 90274-5 . PMID 6456380 .
- ^ Xiong J (2006). Основная биоинформатика . Кембридж, Великобритания: издательство Кембриджского университета. с. 4 . ISBN 978-0-511-16815-4 - через интернет -архив.
- ^ Sanger F, Air GM, Barrell BG, Brown NL, Coulson AR, Fiddes CA, et al. (Февраль 1977 г.). «Нуклеотидная последовательность бактериофага ДНК PHI X174». Природа . 265 (5596): 687–95. Bibcode : 1977natur.265..687s . doi : 10.1038/265687a0 . PMID 870828 . S2CID 4206886 .
- ^ Бенсон Д.А., Карш-Мизрачи И., Липман Д.Дж., Остелл Дж., Уилер Д.Л. (январь 2008 г.). "GenBank" . Исследование нуклеиновых кислот . 36 (проблема базы данных): D25-30. doi : 10.1093/nar/gkm929 . PMC 2238942 . PMID 18073190 .
- ^ Jump up to: а беременный в Fleischmann RD, Adams MD, White O, Clayton RA, Kirkness EF, Kerlavage AR, et al. (Июль 1995 г.). «Случайное секвенирование всего генома и сборка Haemophilus influenzae Rd». Наука . 269 (5223): 496–512. Bibcode : 1995sci ... 269..496f . doi : 10.1126/science.7542800 . PMID 7542800 .
- ^ Stein L (2001). «Аннотация генома: от последовательности к биологии». Природа . 2 (7): 493–503. doi : 10.1038/35080529 . PMID 11433356 . S2CID 12044602 .
- ^ Erdin S, Lisewski AM, Lichtarge O (апрель 2011 г.). «Прогнозирование функции белка: к интеграции метрик сходства» . Современное мнение в структурной биологии . 21 (2): 180–8. doi : 10.1016/j.sbi.2011.02.001 . PMC 3120633 . PMID 21353529 .
- ^ Карваджал-Родригес A (март 2010 г.). «Моделирование генов и геномов вперед во времени» . Текущая геномика . 11 (1): 58–61. doi : 10.2174/138920210790218007 . PMC 2851118 . PMID 20808525 .
- ^ Браун Т.А. (2002). «Мутация, восстановление и рекомбинация». Геномы (2 -е изд.). Манчестер (Великобритания): Оксфорд.
- ^ Картер Н.П., Фиглер Х., Пайпер Дж (октябрь 2002 г.). «Сравнительный анализ сравнительных технологий микрочипов геномной гибридизации: отчет о семинаре, спонсируемом The Wellcome Trust». Цитометрия . 49 (2): 43–8. doi : 10.1002/cyto.10153 . PMID 12357458 .
- ^ Чаудхари Н.М., Гупта В.К., Датта С (апрель 2016 г.). «BPGA-ультрабычный конвейер пан-геном» . Научные отчеты . 6 : 24373. BIBCODE : 2016NATSR ... 624373C . doi : 10.1038/srep24373 . PMC 4829868 . PMID 27071527 .
- ^ Aston Ki (май 2014). «Генетическая восприимчивость к мужскому бесплодию: новости из исследований в области ассоциаций по всему геному» . Андрология . 2 (3): 315–21. doi : 10.1111/j.2047-2927.2014.00188.x . PMID 24574159 . S2CID 206007180 .
- ^ Véron A, Blein S, Cox DG (2014). «Исследования ассоциаций по всему геному и клиника: акцент на рак молочной железы». Биомаркеры в медицине . 8 (2): 287–96. doi : 10.2217/bmm.13.121 . PMID 24521025 .
- ^ Tosto G, Reitz C (октябрь 2013 г.). «Исследования ассоциации по всему геному при болезни Альцгеймера: обзор» . Текущая неврология и неврологические отчеты . 13 (10): 381. doi : 10.1007/s11910-013-0381-0 . PMC 3809844 . PMID 23954969 .
- ^ Лондин Е., Ядав П., Суррей С., Крика Л.Дж., Фортина П. (2013). «Использование анализа связей, исследования ассоциаций по всему геному и секвенирование следующего поколения при идентификации мутаций, вызывающих заболевание». Фармакогеномика . Методы в молекулярной биологии. Тол. 1015. С. 127–46. doi : 10.1007/978-1-62703-435-7_8 . ISBN 978-1-62703-434-0 Полем PMID 23824853 .
- ^ Hendorff LA, Sethupathy P, Junkins HA, Ramos EM, Mehta JP, Collins FS, et al. (Июнь 2009 г.). «Потенциальные этиологические и функциональные последствия локусов ассоциации всего генома для болезней и признаков человека» . Труды Национальной академии наук Соединенных Штатов Америки . 106 (23): 9362–7. Bibcode : 2009pnas..106.9362H . doi : 10.1073/pnas.0903103106 . PMC 2687147 . PMID 19474294 .
- ^ Холл Ло (2010). «Поиск правильных генов для прогнозирования заболеваний и прогноза». 2010 Международная конференция по системной науке и технике . С. 1–2. doi : 10.1109/icsse.2010.5551766 . ISBN 978-1-4244-6472-2 Полем S2CID 21622726 .
- ^ Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hendorff LA, Hunter DJ, et al. (Октябрь 2009 г.). «Поиск пропавшей наследуемости сложных заболеваний» . Природа . 461 (7265): 747–753. Bibcode : 2009natur.461..747m . doi : 10.1038/nature08494 . PMC 2831613 . PMID 19812666 .
- ^ Wainschtein P, Jain D, Zheng Z, Aslibekyan S, Becker D, Bi W, et al. (Март 2022 г.). «Оценка вклада редких вариантов в сложную черту, наследуемость из данных последовательности всего генома» . Природа генетика . 54 (3): 263–273. doi : 10.1038/s41588-021-00997-7 . PMC 9119698 . PMID 35256806 .
- ^ Taliun D, Harris DN, Kessler MD, Carlson J, Szpiech Za, Torres R, et al. (Февраль 2021 г.). «Секвенирование 53 831 разнообразных геномов из программы NHLBI Topmed» . Природа . 590 (7845): 290–299. Bibcode : 2021natur.590..290t . doi : 10.1038/s41586-021-03205-y . PMC 7875770 . PMID 33568819 .
- ^ Li X, Li Z, Zhou H, Gaynor SM, Liu Y, Chen H, et al. (Сентябрь 2020 г.). «Динамическое включение множественных функциональных аннотаций Silico дает возможность анализу редких вариантов ассоциации крупных исследований секвенирования всего генома в масштабе» . Природа генетика . 52 (9): 969–983. doi : 10.1038/s41588-020-0676-4 . PMC 7483769 . PMID 32839606 .
- ^ Li Z, Li X, Zhou H, Gaynor SM, Selvaraj MS, Arapoglou T, et al. (Декабрь 2022 г.). «Структура для обнаружения некодирующих редко-вариантов ассоциаций крупномасштабных исследований секвенирования всего генома» . Природные методы . 19 (12): 1599–1611. doi : 10.1038/s41592-022-01640-x . PMC 10008172 . PMID 36303018 . S2CID 243873361 .
- ^ «Staarpipeline: все в одном редко-вариантном инструменте для данных секвенирования цельного генома в масштабе биобанка». Природные методы . 19 (12): 1532–1533. Декабрь 2022 года. DOI : 10.1038/S41592-022-01641-W . PMID 36316564 . S2CID 253246835 .
- ^ Li X, Quick C, Zhou H, Gaynor SM, Liu Y, Chen H, et al. (Январь 2023 г.). «Мощный, масштабируемый и эффективный ресурсный метаанализ редких вариантов ассоциаций в больших исследованиях секвенирования цельного генома» . Природа генетика . 55 (1): 154–164. doi : 10.1038/s41588-022-01225-6 . PMC 10084891 . PMID 36564505 . S2CID 255084231 .
- ^ Васкес М., Де Ла Торре В., Валенсия А (27 декабря 2012 г.). «Глава 14: Анализ генома рака» . PLOS Computational Biology . 8 (12): E1002824. BIBCODE : 2012PLSCB ... 8E2824V . doi : 10.1371/journal.pcbi.1002824 . PMC 3531315 . PMID 23300415 .
- ^ Hye-Jung EC, Jaswinder K, Martin K, Samuel AA, Marco AM (2014). «Секвенирование второго поколения для анализа генома рака». В Dellaire G, Berman JN, Arceci RJ (Eds.). Геномика рака . Бостон (США): Академическая пресса. С. 13–30. doi : 10.1016/b978-0-12-396967-5.00002-5 . ISBN 978-0-12-396967-5 .
- ^ Grau J, Ben-Gal I, Posch S, Grosse I (июль 2006 г.). «Vombat: прогнозирование сайтов связывания транскрипционного фактора с использованием байесовских деревьев переменного порядка» . Исследование нуклеиновых кислот . 34 (Проблема веб-сервера): W529-33. doi : 10.1093/nar/gkl212 . PMC 1538886 . PMID 16845064 .
- ^ «Атлас белка человека» . www.proteinatlas.org . Архивировано из оригинала 4 марта 2020 года . Получено 2 октября 2017 года .
- ^ «Человеческая клетка» . www.proteinatlas.org . Архивировано с оригинала 2 октября 2017 года . Получено 2 октября 2017 года .
- ^ Thul PJ, Åkesson L, Wiking M, Mahdessian D, Geladaki A, Ait Blal H, et al. (Май 2017). «Субклеточная карта протеома человека». Наука . 356 (6340): EAAL3321. doi : 10.1126/science.aal3321 . PMID 28495876 . S2CID 10744558 .
- ^ Ay F, Noble WS (сентябрь 2015 г.). «Методы анализа для изучения трехмерной архитектуры генома» . Биология генома . 16 (1): 183. doi : 10.1186/s13059-015-0745-7 . PMC 4556012 . PMID 26328929 .
- ^ Kryshtafovych A, Schwede T, Topf M, Fidelis K, Moult J (2019). «Критическая оценка методов прогнозирования структуры белка (CASP) - раунд XIII» . Белки . 87 (12): 1011–1020. doi : 10.1002/prot.25823 . PMC 6927249 . PMID 31589781 .
- ^ "Дом - CASP14" . PredictionCenter.org . Архивировано из оригинала 30 января 2023 года . Получено 12 июня 2023 года .
- ^ Хой JA, Robinson H, Trent JT, Kakar S, Smagghe BJ, Hargrove MS (август 2007 г.). «Растительные гемоглобины: молекулярная ископаемая запись для эволюции транспорта кислорода». Журнал молекулярной биологии . 371 (1): 168–79. doi : 10.1016/j.jmb.2007.05.029 . PMID 17560601 .
- ^ Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, et al. (Август 2021 г.). «Высокий точный прогноз структуры белка с помощью алфалолда» . Природа . 596 (7873): 583–589. Bibcode : 2021natur.596..583j . doi : 10.1038/s41586-021-03819-2 . ISSN 1476-4687 . PMC 8371605 . PMID 34265844 .
- ^ «База данных структуры алфалолдного белка» . alphafold.ebi.ac.uk . Архивировано из оригинала 24 июля 2021 года . Получено 10 октября 2022 года .
- ^ Тиц Б., Раджагопала С.В., Голл Дж., Хаузер Р., Маккевитт М.Т., Палцкилл Т. и др. (Май 2008 г.). Зал N (ред.). «Бинарный белковой взаимодействие Treponema pallidum-Syphilis Spirochete» . Plos один . 3 (5): E2292. Bibcode : 2008ploso ... 3.2292t . doi : 10.1371/journal.pone.0002292 . PMC 2386257 . PMID 18509523 .
- ^ Jump up to: а беременный «Открытый фонд биоинформатики: о нас» . Официальный сайт . Открытый фонд биоинформатики . Архивировано из оригинала 12 мая 2011 года . Получено 10 мая 2011 года .
- ^ «Открытый фонд биоинформатики: Bosc» . Официальный сайт . Открытый фонд биоинформатики . Архивировано из оригинала 18 июля 2011 года . Получено 10 мая 2011 года .
- ^ Нисбет Р., Старейшина IV J, Miner G (2009). «Биоинформатика» . Справочник по статистическому анализу и приложениям для интеллектуального анализа данных . Академическая пресса. п. 328. ISBN 978-0-08-091203-5 .
- ^ Управление комиссара. «Продвижение нормативной науки - 24–25 сентября 2014 г. Публичный семинар: стандарты секвенирования следующего поколения» . www.fda.gov . Архивировано с оригинала 14 ноября 2017 года . Получено 30 ноября 2017 года .
- ^ Simonyan V, Goecks J, Mazumder R (2017). «Биокомпьюте объекты-шаг к оценке и валидации биомедицинских научных вычислений» . КПК Журнал фармацевтической науки и техники . 71 (2): 136–146. doi : 10.5731/pdajpst.2016.006734 . PMC 5510742 . PMID 27974626 .
- ^ Управление комиссара. «Продвижение нормативно-исследовательской науки-развитие стандартов HTS для проверки данных и вычислений и поощрения взаимодействия» . www.fda.gov . Архивировано с оригинала 26 января 2018 года . Получено 30 ноября 2017 года .
- ^ Alterovitz G, Dean D, Goble C, Crusoe MR, Soil и Ryees S, Bell A, et al. (Декабрь 2018). «Обеспечение точной медицины посредством стандартной связи провенанса, анализа и результатов HTS» . PLOS Биология . 16 (12): E3000099. doi : 10.1371/journal.pbio.3000099 . PMC 6338479 . PMID 30596645 .
- ^ Проект BioCompute Object (BCO)-это совместная и управляемая сообществом структуру для стандартизации вычислительных данных HTS. 1. Документ о спецификации BCO: Руководство пользователя для понимания и создания B. , Биокомпублирующие объекты, 3 сентября 2017 года, архивировано с оригинала 27 июня 2018 года , получено 30 ноября 2017 года.
- ^ Кэмпбелл А.М. (1 июня 2003 г.). «Общественный доступ для преподавания геномики, протеомики и биоинформатики» . Клеточная биология образование . 2 (2): 98–111. doi : 10.1187/cbe.03-02-0007 . PMC 162192 . PMID 12888845 .
- ^ Аренас М (сентябрь 2021 г.). «Общие соображения для онлайн -практики преподавания в биоинформатике во времена Covid -19» . Биохимия и молекулярное биологическое образование . 49 (5): 683–684. doi : 10.1002/bmb.21558 . ISSN 1470-8175 . PMC 8426940 . PMID 34231941 .
- ^ Barker D, Ferrier DE, Holland PW, Mitchell JB, Plaisier H, Ritchie MG, et al. (Август 2013). «4273π: Обучение биоинформатике по недорогим оборудованию ARM» . BMC Bioinformatics . 13 : 522. DOI : 10.1186/1471-2105-14-243 . PMC 3751261 . PMID 23937194 .
- ^ Barker D, Alderson RG, McDonagh JL, Plaisier H, Comrie MM, Duncan L, et al. (2015). «Практическая деятельность на уровне университета в биоинформатике приносит пользу добровольным группам учеников за последние 2 года школы» . Международный журнал обучения STEM . 2 (17). doi : 10.1186/s40594-015-0030-z . HDL : 10023/7704 . S2CID 256396656 .
- ^ McDonagh JL, Barker D, Alderson RG (2016). «Предоставление вычислительной науки для общественности» . Springerplus . 5 (259): 259. doi : 10.1186/s40064-016-1856-7 . PMC 4775721 . PMID 27006868 .
- ^ Робсон Дж. Ф., Баркер Д. (октябрь 2015 г.). «Сравнение содержания генов, кодирующего белок, в хламидиозе Trachomatis и Protochlamydia amoebophila с использованием компьютера Raspberry Pi» . BMC Research Notes . 8 (561): 561. DOI : 10.1186/S13104-015-1476-2 . PMC 4604092 . PMID 26462790 .
- ^ Wreggelsworth KM, Barker D (октябрь 2015 г.). «Сравнение кодирующих белок геномов двух зеленых бактерий серы, хлорбиума Tepidum TLS и Pelodictyon phaeoclathratiforme bu-1» . BMC Research Notes . 8 (565): 565. DOI : 10.1186/S13104-015-1535-8 . PMC 4606965 . PMID 26467441 .
Дальнейшее чтение
[ редактировать ]- Sehgal et al. : Структурные, филогенетические и стыковочные исследования активатора оксидазы D-аминокислоты (DAOA), гена кандидата шизофрении. Теоретическая биология и медицинское моделирование 2013 10: 3.
- Achuthsankar S Nair Computational Biology & Bioinformatics - нежный обзор, архивный 16 декабря 2008 года на машине Wayback , Коммуникации Компьютерного общества Индии, январь 2007 г.
- Aluru, Srinivas , ed. Справочник по вычислительной молекулярной биологии . Chapman & Hall/CRC, 2006. ISBN 1-58488-406-1 (серия Chapman & Hall/CRC Computer and Information Science)
- Baldi, P и Brunak, S, Bioinformatics: подход машинного обучения , 2 -е издание. MIT Press, 2001. ISBN 0-262-02506-X
- Barnes, MR и Grey, IC, Eds., Биоинформатика для генетиков , первое издание. Wiley, 2003. ISBN 0-470-84394-2
- Baxevanis, Ad and Ouellette, Bff, Eds., Bioinformatics: практическое руководство по анализу генов и белков , третье издание. Wiley, 2005. ISBN 0-471-47878-4
- Baxevanis, AD, Petsko, GA, Stein, LD и Stormo, GD, Eds., Текущие протоколы в биоинформатике . Wiley, 2007. ISBN 0-471-25093-7
- Cristianini, N. and Hahn, M. Введение в вычислительную геномику архивировала 4 января 2009 года в The Wayback Machine , издательство Cambridge University Press, 2006. ( ISBN 9780521671910 | ISBN 0-521-67191-4 )
- Дурбин Р., С. Эдди, А. Крог и Г. Митчисон, Анализ биологической последовательности . Издательство Кембриджского университета, 1998. ISBN 0-521-62971-3
- Гилберт Д. (сентябрь 2004 г.). «Биоинформатические программные ресурсы» . Брифинги в биоинформатике . 5 (3): 300–4. doi : 10.1093/bib/5.3.300 . PMID 15383216 .
- Keedwell E., Интеллектуальная биоинформатика: применение методов искусственного интеллекта к проблемам биоинформатики . Wiley, 2005. ISBN 0-470-02175-6
- Kohane, et al. Микрочипы для интегративной геномики. MIT Press, 2002. ISBN 0-262-11271-X
- Lund, O. et al. Иммунологическая биоинформатика. MIT Press, 2005. ISBN 0-262-12280-4
- Пахтер, Лиор и Штурмфелс, Бернд . «Алгебраическая статистика для вычислительной биологии» издательство Кембриджского университета, 2005. ISBN 0-521-85700-7
- Pevzner, Pavel A. Вычислительная молекулярная биология: алгоритмический подход The Mit Press, 2000. ISBN 0-262-16197-4
- SOINOV, L. Биоинформатика и распознавание схем объединяются в архивированные 10 мая 2013 года в журнале Wayback Machine Journal of Pattern Research ( JPRR Archived 8 сентября 2008 года на The Wayback Machine ), том 1 (1) 2006 с. 37–41
- Стивенс, Халлам, Life Out of Sequence: история биоинформатики, управляемая данными , Чикаго: Университет Чикагской Прессы, 2013, ISBN 9780226080208
- Тисдалл, Джеймс. «Начало Perl для биоинформатики» О'Рейли, 2001. ISBN 0-596-00080-4
- Катализируя расследование на границе раздела вычисления и биологии (2005). Отчет CSTB Архивировал 28 января 2007
- Расчет секретов жизни: вклад математических наук и вычислений в молекулярную биологию (1995) Архивировано 6 июля 2008 г. на машине Wayback
- Основы вычислительной и системной биологии курс MIT
- Вычислительная биология: геномы, сети, эволюция бесплатного курса MIT Архивировано 8 апреля 2013 года на машине Wayback
