Болезнь Шарко-Мари-Тута
| Болезнь Шарко-Мари-Тута | |
|---|---|
| Другие имена | Нейропатия Шарко-Мари-Тута, атрофия малоберцовых мышц, синдром Дежерина-Соттаса. |
 | |
| Стопа человека с болезнью Шарко-Мари-Тута: отсутствие мышц, высокий свод и когтистые пальцы являются признаками этого генетического заболевания. | |
| Произношение | |
| Специальность | Неврология , подиатрия , ортопедия , физическая медицина и реабилитация |
| Симптомы | Свисание стопы , молоткообразный палец периферических , атрофия мышц голеней и предплечий/кистей. |
| Обычное начало | Детство – ранняя взрослая жизнь |
| Продолжительность | Пожизненный |
| Причины | Семейный анамнез (генетика) |
| Факторы риска | Семейный анамнез (генетика), стопы с высоким сводом , стопы с плоским сводом. |
| Метод диагностики | Генетическое тестирование , исследование нервной проводимости или электромиограмма (ЭМГ) |
| Дифференциальный диагноз | Мышечная дистрофия |
| Уход | Управление для поддержания функции |
| Прогноз | Прогрессивный |
| Частота | Распространенность: 1 на 2500 [1] [2] |
Болезнь Шарко-Мари-Тута ( ШМТ ) — наследственная моторная и сенсорная нейропатия периферической нервной системы, характеризующаяся прогрессирующей потерей мышечной ткани и чувствительности к прикосновению в различных частях тела. Это заболевание является наиболее частым наследственным неврологическим расстройством , которым страдает примерно один из 2500 человек. [3] [4] Он назван в честь тех, кто его классически описал: француза Жана-Мартена Шарко (1825–1893), его ученика Пьера Мари (1853–1940), [5] и британец Говард Генри Тут (1856–1925). [6] [7]
Не существует известного лечения. Уход направлен на поддержание функции. ШМТ ранее классифицировалась как подтип мышечной дистрофии . [3]
Признаки и симптомы
[ редактировать ]Симптомы ШМТ обычно начинаются в раннем детстве или раннем взрослом возрасте, но могут появиться и позже. Некоторые люди не испытывают симптомов до 30-40 лет. Обычно первоначальным симптомом является свисание стопы или высокий свод стопы на ранних стадиях заболевания. Это может сопровождаться молоткообразными пальцами , при которых пальцы ног всегда согнуты. Истощение мышечной ткани нижних частей ног может привести к появлению «аистовой ноги» или «перевернутой бутылки шампанского». Слабость в руках и предплечьях возникает у многих людей по мере прогрессирования заболевания. [8]
Потеря осязания в стопах, лодыжках и голенях, а также в кистях, запястьях и руках встречается при различных формах заболевания. Формы с ранним и поздним началом возникают с периодическими болезненными спазматическими мышечными сокращениями, которые могут привести к инвалидности при активации заболевания. Стопы с высоким сводом ( pes cavus ) или стопы с плоским сводом ( pes planus ) классически связаны с этим заболеванием. [9] Сенсорные и проприоцептивные нервы кистей и стоп часто повреждаются, тогда как немиелиновые болевые нервы остаются неповрежденными. Чрезмерное использование пораженной руки или конечности может активировать такие симптомы, как онемение, спазм и болезненные судороги. [8]
Симптомы и течение заболевания могут различаться. Непроизвольное скрежетание зубами и косоглазие широко распространены и часто остаются незамеченными для пострадавшего. У некоторых может быть нарушено дыхание, а также слух, зрение, мышцы шеи и плеч. Сколиоз является распространенным явлением, вызывающим сгорбленность и потерю роста. Тазобедренные суставы могут быть деформированы. Желудочно-кишечные проблемы могут быть частью ШМТ. [10] [11] а также трудности с жеванием, глотанием и речью (из-за атрофии голосовых связок ). [12] Тремор . может развиться по мере истощения мышц беременность Известно, что усугубляет ШМТ, а также сильный эмоциональный стресс. Пациентам с ШМТ следует избегать периодов длительной неподвижности, например, при восстановлении после вторичной травмы, поскольку длительные периоды ограничения подвижности могут резко ускорить симптомы ШМТ. [13]
Боль из-за изменений осанки, скелетных деформаций, мышечной усталости и спазмов довольно часто встречается у людей с ШМТ. Его можно смягчить или вылечить с помощью физиотерапии, хирургического вмешательства, а также корректирующих или вспомогательных устройств. Анальгетики также могут потребоваться, если другие методы лечения не облегчают боль. [14] Нейропатическая боль часто является симптомом ШМТ, хотя, как и другие симптомы ШМТ, ее наличие и тяжесть варьируются от случая к случаю. У некоторых людей боль может быть значительной или сильной и мешать повседневной жизни. Однако не все люди с ШМТ испытывают боль. Когда нейропатическая боль присутствует как симптом ШМТ, она сравнима с болью, наблюдаемой при других периферических невропатиях , а также постгерпетической невралгии и комплексном регионарном болевом синдроме , среди других заболеваний. [15]
Причины
[ редактировать ]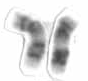
Болезнь Шарко-Мари-Тута вызвана генетическими мутациями , которые вызывают дефекты в нейрональных белках. Нервные сигналы передаются аксоном , окруженным . миелиновой оболочкой Большинство мутаций ШМТ затрагивают миелиновую оболочку, но некоторые затрагивают аксон. [16]
Классификация
[ редактировать ]ШМТ — гетерогенное заболевание , и связанные с ним мутации могут возникать в ряде различных генов. [17] В зависимости от пораженного гена ШМТ подразделяют на несколько типов и подтипов. [18]
Хромосома 17
[ редактировать ]Наиболее частой причиной ШМТ (70–80% случаев) является дупликация большого участка короткого плеча 17-й хромосомы , включающего ген PMP22 . [19]
Хромосома 1
[ редактировать ]Некоторые мутации затрагивают ген MFN2 на хромосоме 1 , который кодирует митохондриальный белок. Мутировавший MFN2 заставляет митохондрии образовывать большие кластеры или сгустки, которые не могут перемещаться по аксону к синапсам . Это препятствует функционированию синапсов. [20]
Х-сцепленные клетки ШМТ и Шванна
[ редактировать ]ШМТ также может быть вызвана Х-сцепленными мутациями и называется Х-сцепленной ШМТ (CMTX). В CMTX мутировавшие коннексоны создают нефункциональные щелевые соединения, которые прерывают молекулярный обмен и транспортировку сигналов. [21] [22] [23]
Мутация может появиться в гене GJB1, кодирующем белок коннексин 32 , белок щелевого соединения, экспрессируемый в шванновских клетках. Поскольку этот белок также присутствует в олигодендроцитах , в ЦНС может возникнуть демиелинизация. [24]
Шванновские клетки создают миелиновую оболочку, обертывая свою плазматическую мембрану вокруг аксона. [21]
Нейроны, шванновские клетки и фибробласты работают вместе, создавая функциональный нерв. Шванновские клетки и нейроны обмениваются молекулярными сигналами через щелевые соединения , которые регулируют выживание и дифференцировку. [25]
Демиелинизирующие шванновские клетки вызывают аномальную структуру и функцию аксонов. Они могут вызвать дегенерацию аксонов или просто привести к их неисправности. [3]
Миелиновая оболочка позволяет нервным клеткам быстрее проводить сигналы. Когда миелиновая оболочка повреждена, нервные сигналы замедляются, и это можно измерить с помощью обычного неврологического теста — электромиографии . Однако когда аксон повреждается, это приводит к снижению потенциала действия сложных мышц . [26]
Аксональная нейропатия, связанная с GARS1
Типы ШМТ2 обычно называют аксональными нейропатиями из-за наблюдаемой аксональной дегенерации. Типы ШМТ2 являются результатом повреждения нервных аксонов , а не повреждения миелиновой оболочки (как в случае с ШМТ1). Поврежденные аксоны вызывают замедление передачи сигналов к мышцам и мозгу, вызывая такие симптомы, как атрофия мышц, слабость, снижение чувствительности и деформация стопы. Симптомы типа ШМТ2 обычно появляются в возрасте от 5 до 25 лет. [27] ШМТ2D является одной из 31 формы Шарко-Мари-Тута 2 типа 1 и диагностируется только в том случае, если наряду с двигательными дефицитами наблюдаются сенсорные дефициты (например, потеря чувствительности из-за деградации сенсорных аксонов); в противном случае дистальную наследственную моторную нейропатию V типа диагностируют . Неизвестно, почему сенсорное участие так различается у пациентов с нейропатией GARS1. [28] Симптомы ШМТ2D включают деформацию стопы, мышечную слабость и судороги, нарушение рефлексов, потерю чувствительности и мышечную атрофию и сходны с симптомами других типов ШМТ1 и ШМТ2. Симптомы и степень тяжести варьируются от пациента к пациенту. [29]
Мышей часто используют для моделирования CMT2D, и они обычно демонстрируют аберрантную нервно-мышечную функцию в нервно-мышечном соединении (НМС). [30] [31] [32] У мышей CMT2D нервно-мышечные соединения аномальны, при этом у субъектов наблюдается дегенерация нервно-мышечных соединений в задних мышцах. На ганглии дорсальных корешков (DRG) также влияет аберрантная судьба сенсорных нейронов, что означает, что судьбы клеток сенсорных нейронов определяются аномально. У мышей CMT2D меньше проприоцептивных и механочувствительных нейронов, но больше ноцицептивных нейронов, возможно, из-за мутантного GlyRS, аберрантно взаимодействующего с внеклеточной областью киназных рецепторов тропомиозина, или Trk, рецепторов. [33] Рецепторы Trk имеют решающее значение для выживания и развития сенсорных нейронов; при разрушении также нарушается развитие и выживание нервов, что, возможно, приводит к аномальному количеству сенсорных нейронов, наблюдаемому у мышей CMT2D. [28]
ШМТ2D является результатом аутосомно-доминантной мутации в гене человека GARS1, расположенном в позиции 7p14.3. [34] Считается, что он вызван аберрантными миссенс-мутациями, связанными с усилением функции . [28] Ген GARS1 представляет собой белок-кодирующий ген, ответственный за кодирование глицил-тРНК-синтетазы (GlyRS). класса II Глицил-тРНК-синтетаза представляет собой аминоацил-тРНК-синтетазу и действует как катализатор синтеза глицил-тРНК путем ковалентного связывания аминокислот с соответствующими родственными им тРНК для трансляции белка . Глицил-тРНК-синтетаза является неотъемлемой частью трансляции белка и присоединяет глицин к родственной ему тРНК. [35]
У пациентов с CMT2D было обнаружено множество различных мутаций, и остается неясным, как мутации в GARS1 вызывают CMT2D. Однако считается, что мутантная глицил-тРНК-синтетаза (GlyRS) взаимодействует с трансмембранными рецепторами, вызывая двигательные заболевания. [36] [37] и что мутации в гене могут нарушить способность GlyRS взаимодействовать с родственной ему РНК, нарушая выработку белка. Мутации GARS1, присутствующие в CMT2D, вызывают недостаточное количество глицил-тРНК в клетках, предотвращая фазу элонгации синтеза белка . Поскольку элонгация является ключевым этапом в производстве белка, рибосомы не могут продолжать синтез белка в участках глицина. Мутации GARS1 также останавливают инициацию трансляции. Неудачное добавление глицина вызывает стрессовую реакцию, которая еще больше останавливает выработку белка, предотвращая инициацию трансляции. Задерживая элонгацию и инициацию трансляции, мутации CMT2D в гене GARS1 вызывают репрессию трансляции, а это означает, что трансляция ингибируется в целом. [38]
Аксональная нейропатия, связанная с GARS1, является прогрессирующей , то есть со временем ее состояние ухудшается. Считается, что неизвестные механизмы вызывают хроническую нейродегенерацию , возникающую в результате аберрантного GlyRS; однако одной из теорий заболевания является дефицит VEGF. Мутантный GlysRS взаимодействует с нейрональными трансмембранными рецепторами, включая нейропилин 1 (Nrp1) и фактор роста эндотелия сосудов (VEGF) , вызывая нейропатию. [37] Мутации GARS-CMT2D изменяют GlyRS и позволяют ему связываться с рецептором Nrp1, мешая нормальному связыванию Nrp1 с VEGF. В то время как повышенная экспрессия VEGF улучшает двигательную функцию, снижение экспрессии Nrp1 ухудшает CMT2D; поскольку Nrp1 связывается с мутантным GlyRS у мутантных людей GARS1-CMT2D, экспрессия Nrp1 снижается, что, в свою очередь, ухудшает двигательную функцию. У мышей с дефицитом VEGF со временем наблюдаются заболевания двигательных нейронов. Таким образом, путь VEGF/Nrp1 считается пригодным для лечения CMT2D. [27]
Диагностика
[ редактировать ]ШМТ можно диагностировать с помощью трех различных форм тестов: измерение скорости нервных импульсов ( исследования нервной проводимости ), биопсия нерва и тестирование ДНК. ДНК-тестирование может дать окончательный диагноз, но не все генетические маркеры ШМТ известны. ШМТ в первую очередь наиболее заметна, когда у кого-то развивается слабость голени, например, свисание стопы или деформации стопы, включая молоткообразные пальцы и высокий свод стопы, но сами по себе симптомы не позволяют поставить диагноз. Пациенты должны быть направлены к врачу, специализирующемуся в неврологии или реабилитационной медицине. Чтобы увидеть признаки мышечной слабости, невролог может попросить пациентов походить на пятках или переместить часть ноги против противодействующей силы. Чтобы выявить потерю чувствительности, невролог проверяет глубокие сухожильные рефлексы, такие как коленный рефлекс, которые при ШМТ снижены или отсутствуют. Врач также может узнать семейный анамнез пациента, поскольку ШМТ передается по наследству. Отсутствие семейного анамнеза не исключает ШМТ, но помогает исключить другие причины невропатии, такие как диабет или воздействие определенных химических веществ или лекарств. [39]
В 2010 году ШМТ была одним из первых заболеваний, при котором генетическая причина заболевания конкретного пациента была точно определена путем секвенирования всего генома больного человека. Это сделали ученые, работающие в Ассоциации Шарко Мари Тут (CMTA). [40] [18] Две мутации были идентифицированы в гене SH3TC2 , который, как известно, вызывает ШМТ. Затем исследователи сравнили геном больного пациента с геномами матери, отца и семи братьев и сестер пациента с заболеванием и без него. У матери и отца было по одной нормальной и одной мутантной копии этого гена, а симптомы были легкими или отсутствовали. Потомство, унаследовавшее два мутантных гена, полностью заболело. [18]
Гистология
[ редактировать ]
Постоянный цикл демиелинизации и ремиелинизации , который происходит при ШМТ, может привести к образованию слоев миелина вокруг некоторых нервов, называемых «луковой луковицей». Они также наблюдаются при хронической воспалительной демиелинизирующей полинейропатии . [41] В мышцах наблюдается группировка волокон по типам — аналогичный неспецифический признак, указывающий на цикл денервации / реиннервации . Обычно мышечные волокна типа I и типа II имеют случайное распределение в виде шахматной доски. Однако при реиннервации группы волокон, связанные с одним нервом, являются однотипными. Стандартом для определения типа клетчатки является гистоферментативная аденозинтрифосфатаза (АТФаза при pH 9,4). [42]
Управление
[ редактировать ]Часто наиболее важной целью пациентов с ШМТ является поддержание движения, мышечной силы и гибкости. Поэтому рекомендуется подход межпрофессиональной команды с участием трудотерапии (ОТ), физиотерапии (ПТ), ортопеда, ортопеда и/или хирурга-ортопеда. [8]
Подходящая обувь также очень важна для людей с ШМТ, но им часто трудно найти подходящую обувь из-за высокого свода стопы и молоткообразных пальцев ног. Из-за отсутствия хорошей сенсорной рецепции в стопах пациентам с ШМТ также может потребоваться посещение ортопеда для стрижки ногтей или удаления мозолей, образующихся на подушечках стоп. Наконец, пациенты также могут принять решение о проведении операции у ортопеда или хирурга-ортопеда. Хирургическое вмешательство может помочь стабилизировать стопы пациентов или исправить прогрессирующие проблемы. Эти процедуры включают выпрямление и фиксацию пальцев ног, опускание свода стопы, а иногда и сращивание голеностопного сустава для обеспечения устойчивости. [13] Пациенты с ШМТ должны проявлять особую осторожность, чтобы избежать падения, поскольку у людей с основным заболеванием заживление переломов занимает больше времени. Кроме того, возникающее в результате бездействие может привести к ухудшению ШМТ. [13] Ассоциация Шарко-Мари-Тута классифицирует химиотерапевтический препарат винкристин как препарат «определенно высокого риска» и заявляет, что «винкристин оказался опасным, и его следует избегать всем пациентам с ШМТ, включая тех, у кого нет симптомов». [43] Для улучшения физического состояния пострадавших можно провести несколько корректирующих хирургических процедур. [44]
Ортопедические стельки
[ редактировать ]
Если мышцы нижних конечностей слабые, имеет смысл назначить ортопедические стельки индивидуального изготовления . В зависимости от того, какие группы мышц поражены, следует назначать правильные ортезы с соответствующими функциональными элементами. Слабость передней большеберцовой мышцы , поднимающей стопу, обычно сопровождается атрофией икроножной мышцы , которая вместе с камбаловидной мышцей образует трехглавую мышцу голени (дистальную часть икроножной мышцы), вызывая известную «деформацию ноги аиста». ". [45] В большинстве случаев имеют смысл использовать голеностопные ортезы , имеющие функциональные элементы подъема стопы и регулируемый контроль опускания переднего отдела стопы. Слабые икроножные мышцы приводят к недостаточной активации рычага передней части стопы. Это приводит к дополнительному увеличению неуверенности при стоянии и ходьбе. Если икроножные мышцы слабы, ортез должен быть оснащен функциональными элементами для активации рычага передней части стопы. Для этого рекомендуется ортопедический сустав с регулируемым динамическим стопором тыльного сгибания с мощной пружиной в сочетании с кожухом голени перед голенью. Такие ортезы помогают контролировать свисание стопы, нестабильность стопы и голеностопного сустава, а также обеспечивают пациенту лучшее чувство равновесия при стоянии и ходьбе, не ограничивая подвижность и динамику голеностопного сустава. Исследования подтверждают положительный эффект ортезов с регулируемыми функциональными элементами у больных с параличом этих групп мышц. [46] [47] [48] [49] Большим преимуществом является то, что сопротивления двух функциональных элементов могут быть установлены отдельно друг от друга в двух направлениях движения: тыльном сгибании и подошвенном сгибании . [50]
Прогноз
[ редактировать ]Тяжесть симптомов широко варьируется даже для одного и того же типа ШМТ. случаях монозиготных близнецов Сообщалось о с разной степенью тяжести заболевания, что показывает, что идентичные генотипы связаны с разными уровнями тяжести заболевания (см. Пенетрантность ). Некоторые пациенты могут жить нормальной жизнью и почти или полностью не имеют симптомов. [51] В обзоре 2007 года говорится, что «неизвестно, что ожидаемая продолжительность жизни в большинстве случаев изменилась». [52]
История
[ редактировать ]


Заболевание названо в честь тех, кто его классически описал: француза Жана-Мартена Шарко (1825–1893), его ученика Пьера Мари (1853–1940), [5] и британец Говард Генри Тут (1856–1925). [6]
См. также
[ редактировать ]- Классификации болезней Шарко-Мари-Тута
- Ладонно-подошвенная кератодермия и спастическая параплегия.
- Наследственные моторные и сенсорные нейропатии
- Наследственные моторные невропатии
- Низкое количество повторов копии
- Мир Кристины
Ссылки
[ редактировать ]- ^ Корнетт К.М., Менезес М.П., Брей П., Халаки М., Шай Р.Р., Юм С.В. и др. (июнь 2016 г.). «Фенотипическая изменчивость детской болезни Шарко-Мари-Тута» . JAMA Неврология . 73 (6): 645–651. дои : 10.1001/jamaneurol.2016.0171 . ПМЦ 4916861 . ПМИД 27043305 .
- ^ Скре Х (23 апреля 2008 г.). «Генетические и клинические аспекты болезни Шарко-Мари-Тута». Клиническая генетика . 6 (2): 98–118. дои : 10.1111/j.1399-0004.1974.tb00638.x . ПМИД 4430158 . S2CID 45225191 .
- ^ Jump up to: а б с Краевски К.М., Льюис Р.А., Фюрст Д.Р., Туранский С., Хиндерер С.Р., Гарберн Дж. и др. (июль 2000 г.). «Неврологическая дисфункция и аксональная дегенерация при болезни Шарко-Мари-Тута типа 1А» . Мозг . 123 (7): 1516–1527. дои : 10.1093/мозг/123.7.1516 . ПМИД 10869062 .
- ^ «Физическая медицина и реабилитация при болезни Шарко-Мари-Тута: предыстория, патофизиология, эпидемиология» . Emedicine.medscape.com . Проверено 13 ноября 2016 г.
- ^ Jump up to: а б Шарко Ж.М. (1886). «Об особой форме прогрессирующей мышечной атрофии, часто семейной, начинающейся со стоп и ног и позднее достигающей рук». Медицинский обзор (на французском языке). 6 :97–138.
- ^ Jump up to: а б Зуб НХ (1886 г.). Перонеальный тип прогрессирующей мышечной атрофии (диссертация доктора медицинских наук). Лондон.
- ^ Кумар Д.Р., Аслиния Ф., Йельский университет Ш., Мацца Дж.Дж. (март 2011 г.). «Жан-Мартен Шарко: отец неврологии» . Клиническая медицина и исследования . 9 (1): 46–49. дои : 10.3121/cmr.2009.883 . ISSN 1539-4182 . ПМК 3064755 . ПМИД 20739583 .
- ^ Jump up to: а б с «Информационный бюллетень о болезни Шарко-Мари-Тута» . Национальный институт неврологических расстройств и инсульта . Проверено 23 апреля 2022 г.
- ^ Ле Т, Бхушан В (6 января 2014 г.). Первая помощь для USMLE, шаг 1, 2014 г. Макгроу-Хилл Образование. ISBN 978-0-07-183142-0 . Проверено 4 сентября 2014 г.
Обычно аутосомно-доминантный тип наследования связан со сколиозом и деформациями стопы (высокий или плоский свод).
- ^ «Новости СМТ» . Lindacrabtree.com . Архивировано из оригинала 5 августа 2016 г. Проверено 13 ноября 2016 г.
- ^ Сойкан I, МакКаллум RW (январь 1997 г.). «Вовлечение желудочно-кишечного тракта при неврологических расстройствах: синдромы Стиффмана и Шарко-Мари-Тута». Американский журнал медицинских наук . 313 (1): 70–73. дои : 10.1097/00000441-199701000-00012 . ПМИД 9001170 .
- ^ «Информационный бюллетень о болезни Шарко-Мари-Тута» . Национальный институт неврологических расстройств и инсульта . 14 января 2016 г. Архивировано из оригинала 19 ноября 2016 г. Проверено 13 ноября 2016 г.
- ^ Jump up to: а б с «Лечение и ведение ШМТ» (Пресс-релиз). Ассоциация Шарко-Мари-Тута. 6 октября 2010 г. Проверено 26 августа 2011 г.
- ^ «Синдром Шарко-Мари-Тута. Информация о ШМТ» . Пациент . 20 августа 2021 г.
- ^ Картер Г.Т., Дженсен М.П., Галер Б.С., Крафт Г.Х., Крэбтри Л.Д., Бердсли Р.М. и др. (декабрь 1998 г.). «Нейропатическая боль при болезни Шарко-Мари-Тута». Архив физической медицины и реабилитации . 79 (12): 1560–1564. дои : 10.1016/S0003-9993(98)90421-X . ПМИД 9862301 .
- ^ Ниманн А., Бергер П., Сутер У. (март 2006 г.). «Патомеханизмы мутантных белков при болезни Шарко-Мари-Тута» . Нейромолекулярная медицина . 8 (1–2): 217–241. doi : 10.1385/NMM:8:1-2:217 (неактивен 14 марта 2024 г.). hdl : 20.500.11850/422903 . ISSN 1535-1084 . ПМИД 16775378 . S2CID 17130051 .
{{cite journal}}: CS1 maint: DOI неактивен по состоянию на март 2024 г. ( ссылка ) [ постоянная мертвая ссылка ] - ^ Хойл Дж.К., Исфорт MC, Роггенбак Дж., Арнольд В.Д. (2015). «Генетика болезни Шарко-Мари-Тута: текущие тенденции и будущие последствия для диагностики и лечения» . Применение клинической генетики . 8 : 235–243. дои : 10.2147/TACG.S69969 . ПМК 4621202 . ПМИД 26527893 .
- ^ Jump up to: а б с Лупски Дж.Р., Рид Дж.Г., Гонзага-Хореги С., Рио Дейрос Д., Чен Д.С., Назарет Л. и др. (апрель 2010 г.). «Полногеномное секвенирование у пациента с невропатией Шарко-Мари-Тута» . Медицинский журнал Новой Англии . 362 (13): 1181–1191. doi : 10.1056/NEJMoa0908094 . ПМК 4036802 . ПМИД 20220177 .
- ^ Флореску С., Албу К.В., Думитреску С., Тыртя Г.К., Флореску О.А., Тыртя Е.А. (2017). «Нарушения сна и памяти у больного, страдающего болезнью Шарко-Мари-Тута» . Текущий журнал медицинских наук . 43 (1): 73–77. дои : 10.12865/CHSJ.43.01.11 . ПМК 6286719 . ПМИД 30595858 .
- ^ Балох Р.Х., Шмидт Р.Э., Пестронк А., Милбрандт Дж. (январь 2007 г.). «Измененный аксональный митохондриальный транспорт в патогенезе болезни Шарко-Мари-Тута из-за мутаций митофузина 2» . Журнал неврологии . 27 (2): 422–430. doi : 10.1523/JNEUROSCI.4798-06.2007 . ПМК 6672077 . ПМИД 17215403 .
- ^ Jump up to: а б Бергер П., Янг П., Сутер У (март 2002 г.). «Молекулярно-клеточная биология болезни Шарко-Мари-Тута». Нейрогенетика . 4 (1): 1–15. дои : 10.1007/s10048-002-0130-z . ПМИД 12030326 . S2CID 25129077 .
- ^ Клеопа К.А. (декабрь 2011 г.). «Роль щелевых соединений при болезни Шарко-Мари-Тута» . Журнал неврологии . 31 (49): 17753–17760. doi : 10.1523/JNEUROSCI.4824-11.2011 . ПМК 6634164 . ПМИД 22159091 .
- ^ Сигети К., Лупски-младший (июнь 2009 г.). «Болезнь Шарко-Мари-Тута» . Европейский журнал генетики человека . 17 (6): 703–710. дои : 10.1038/ejhg.2009.31 . ПМК 2947101 . ПМИД 19277060 .
- ^ Кутсис Г., Бреза М., Велонакис Г., Цартос Дж., Касселимис Д., Картану С. и др. (февраль 2019 г.). «X-связанная болезнь Шарко-Мари-Тута и рассеянный склероз: новые доказательства связи». Журнал неврологии, нейрохирургии и психиатрии . 90 (2): 187–194. дои : 10.1136/jnnp-2018-319014 . ПМИД 30196252 . S2CID 52175657 .
- ^ Амиотт Э.А., Лотт П., Сото Дж., Канг П.Б., Маккаффери Дж.М., ДиМауро С. и др. (май 2008 г.). «Слияние и функция митохондрий в фибробластах пациентов Шарко-Мари-Тута типа 2А с мутациями митофузина 2» . Экспериментальная неврология . 211 (1): 115–127. doi : 10.1016/j.expneurol.2008.01.010 . ПМК 2409111 . ПМИД 18316077 .
- ^ Ю Э.М., Бернс Дж., Райан М.М. , Уврие Р.А. (сентябрь 2008 г.). «Нейрофизиологические нарушения у детей с болезнью Шарко-Мари-Тута типа 1А». Журнал периферической нервной системы . 13 (3): 236–241. дои : 10.1111/j.1529-8027.2008.00182.x . ПМИД 18844790 . S2CID 205694771 .
- ^ Jump up to: а б «ШМТ2 — Типы болезни Шарко-Мари-Тута (ШМТ) — Заболевания» . Ассоциация мышечной дистрофии . 23 декабря 2015 г. Проверено 10 мая 2022 г.
- ^ Jump up to: а б с Сани Дж.Н., Мех А.М., Актар Т., Чжан Ю., Скьяво Г. (2020). «Измененное развитие сенсорных нейронов у мышей CMT2D зависит от места и связано с повышенным уровнем GlyRS» . Границы клеточной нейронауки . 14 : 232. дои : 10.3389/fncel.2020.00232 . ПМЦ 7431706 . ПМИД 32848623 .
- ^ «Болезнь Шарко-Мари-Тута, тип 2D (идентификатор концепции: C1832274)» . МедГен — NCBI . Проверено 10 мая 2022 г.
- ^ Сани Дж.Н., Грайс С.Дж., Берджесс Р.В., Талбот К., Кадер М.З. (2014). «Дефекты созревания нервно-мышечных соединений предшествуют нарушению связи нижних мотонейронов у мышей Шарко-Мари-Тута типа 2D» . Хум Мол Жене . 15 (10): 2639–50. дои : 10.1093/hmg/ddt659 . ПМК 3990164 . ПМИД 24368416 .
- ^ Сполдинг Э.Л., Сани Дж.Н., Морелли К.Х., Пинтер М.Дж., Берджесс Р.В., Себерн К.Л. (2016). «Синаптический дефицит в нервно-мышечных соединениях у двух мышей с типом Шарко-Мари-Тута 2d» . Дж. Нейроски . 16 (11): 3254–67. doi : 10.1523/JNEUROSCI.1762-15.2016 . ПМЦ 4792937 . ПМИД 26985035 .
- ^ Сани Дж.Н., Мех AM, Скьяво Дж. (2020). «Требования развития способствуют ранней нервно-мышечной дегенерации у мышей CMT2D» . Смерть клетки Дис . 11 (7): 564. doi : 10.1038/s41419-020-02798-y . ПМЦ 7378196 . ПМИД 32703932 .
- ^ Сани Дж.Н., Дауэс Дж.М., Вест С.Дж., Вэй Н., Сполдинг Э.Л., Гомес-Мартин А. и др. (2017). «Передача сигналов рецептора Trk и судьба сенсорных нейронов нарушаются при нейропатии человека, вызванной мутациями Gars» . Proc Natl Acad Sci США . 114 (16): E3324–E3333. Бибкод : 2017PNAS..114E3324S . дои : 10.1073/pnas.1614557114 . ПМК 5402433 . ПМИД 28351971 .
- ^ «Запись OMIM - № 601472 - Болезнь Шарко-Мари-Тута, аксональная, тип 2D; CMT2D» . Интернет-менделевское наследование у человека . Проверено 10 мая 2022 г.
- ^ «OMIM Entry-*600287 — Гликл-тРНК-синтетаза 1; GARS1» . Интернет-менделевское наследование у человека . Проверено 10 мая 2022 г.
- ^ Вэй Н, Чжан Ц, Ян XL (2019). «Нейродегенеративная болезнь Шарко-Мари-Тута как пример для расшифровки новых функций аминоацил-тРНК-синтетаз» . J Биол Хим . 294 (14): 5321–5339. дои : 10.1074/jbc.REV118.002955 . ПМК 6462521 . ПМИД 30643024 .
- ^ Jump up to: а б Хэ В., Бай Г., Чжоу Х., Вэй Н., Уайт Н.М., Лауэр Дж. и др. (октябрь 2015 г.). «Нейропатия CMT2D связана с неоморфной связывающей активностью глицил-тРНК-синтетазы» . Природа . 526 (7575): 710–714. Бибкод : 2015Natur.526..710H . дои : 10.1038/nature15510 . ПМЦ 4754353 . ПМИД 26503042 .
- ^ Мендонса С., фон Кюгельген Н., Буянич Л., Чекулаева М. (сентябрь 2021 г.). «Мутация Шарко-Мари-Тута в глицил-тРНК-синтетазе останавливает рибосомы в состоянии предварительной аккомодации и активирует интегрированную реакцию на стресс» . Исследования нуклеиновых кислот . 49 (17): 10007–10017. дои : 10.1093/nar/gkab730 . ПМЦ 8464049 . ПМИД 34403468 .
- ^ «Диагностика ШМТ» . Ассоциация Шарко-Мари-Тута . Проверено 30 мая 2020 г.
- ^ Уэйд Н (10 марта 2010 г.). «Причина заболевания определяется геномом» . Нью-Йорк Таймс . Архивировано из оригинала 1 января 2022 г.
- ^ Мидрони Дж., Бильбао Дж. М., Коэн С. М. (1995). Биопсия-диагностика периферической нейропатии . Бостон: Баттерворт-Хайнеманн. стр. 75 –103. ISBN 978-0-7506-9552-7 .
- ^ Дубовиц В., Сьюри Калифорния, Олдфорс А., Лейн Р. (2013). Биопсия мышц: практический подход (Четвертое изд.). Филадельфия: Сондерс/Эльзевир. ISBN 978-0-7020-4340-6 .
- ^ «Медицинская тревога» . Ассоциация Шарко-Мари-Тута . Архивировано из оригинала 2 июля 2007 г. Проверено 21 августа 2007 г.
- ^ Ананд Н., Левин, Д.Б. , Берк С., Бансал М. Нейропатическая спинальная атрофия при болезни Шарко-Мари-Тута. J Bone Joint Surg. 1997 год; 79-А: 1235–39.
- ^ Агирре-Родригес Ф.Д., Лусенилья М.И., Альварес-Куберо М.Дж., Мата К., Энтрала-Берналь К., Фернандес-Росадо Ф. (октябрь 2015 г.). «Новая мутация FA2H у девочки с семейной спастической параплегией» . Журнал неврологических наук . 357 (1–2): 332–334. дои : 10.1016/j.jns.2015.08.1183 . ПМИД 26344562 .
- ^ Кобаяши Т., Люнг А.К., Акадзава Ю., Хатчинс С.В. (март 2013 г.). «Влияние изменения сопротивления подошвенному сгибанию голеностопного ортеза на кинематику коленного сустава у пациентов с инсультом». Походка и осанка . 37 (3): 457–459. дои : 10.1016/j.gaitpost.2012.07.028 . ПМИД 22921491 .
- ^ Мейнс П., Керкум Ю.Л., Брем М.А., Бехер Дж.Г., Бьюзер А.И., Харлаар Дж. (май 2020 г.). «Ортезы голеностопного сустава при церебральном параличе: влияние жесткости голеностопного сустава на кинематику туловища, стабильность походки и затраты энергии на ходьбу» . Европейский журнал детской неврологии . 26 : 68–74. дои : 10.1016/j.ejpn.2020.02.009 . ПМИД 32147412 . S2CID 212641072 .
- ^ Мейнс П., Керкум Ю., Бьюзер А., Бехер Дж., Брем М., Харлаар Дж. (01.09.2016). «Влияние жесткости ортеза голеностопного сустава на движение туловища и затраты энергии при ходьбе при церебральном параличе». Походка и осанка (на немецком языке). 49 . п. 2. дои : 10.1016/j.gaitpost.2016.07.070 . ISSN 0966-6362 .
- ^ Керкум Ю.Л., Бьюзер А.И., ван ден Ноорт Дж.К., Бехер Дж.Г., Харлаар Дж., Брем М.А. (23 ноября 2015 г.). «Влияние различной жесткости ортезов голеностопного сустава на походку у детей со спастическим церебральным параличом, которые ходят с чрезмерным сгибанием коленей» . PLOS ONE (на немецком языке). 10 (11): e0142878. Бибкод : 2015PLoSO..1042878K . дои : 10.1371/journal.pone.0142878 . ПМЦ 4658111 . ПМИД 26600039 .
- ^ Плогер Х.Э., Ватерваль Н.Ф., Ноллет Ф., Бус С.А., Брем М.А. (2019). «Модификация жесткости двух типов ортезов голеностопного сустава для оптимизации походки у людей с неспастической слабостью икроножных мышц — исследование, подтверждающее концепцию» . Журнал исследований стопы и голеностопного сустава (на немецком языке). 12:41 . дои : 10.1186/s13047-019-0348-8 . ПМК 6686412 . ПМИД 31406508 .
- ^ Парейсон Д., Маркези К. (июль 2009 г.). «Диагностика, естественное течение и лечение болезни Шарко-Мари-Тута». «Ланцет». Неврология . 8 (7): 654–667. дои : 10.1016/S1474-4422(09)70110-3 . ПМИД 19539237 . S2CID 665324 .
- ^ Абуссуан Л.С., Льюис Р.А., Шай М.Э. (9 февраля 2007 г.). «Нарушения функции легких, сна и верхних дыхательных путей при болезни Шарко-Мари-Тута». Легкое . 185 (1): 1–7. дои : 10.1007/s00408-006-0053-9 . ПМИД 17294338 . S2CID 12889721 .
Внешние ссылки
[ редактировать ] СМИ, связанные с болезнью Шарко-Мари-Тута, на Викискладе?
СМИ, связанные с болезнью Шарко-Мари-Тута, на Викискладе? - Болезнь Шарко-Мари-Тута в Керли
| Базы данных органов управления : Национальные |
|---|