болезнь Хантингтона
| болезнь Хантингтона | |
|---|---|
| Другие имена | хорея Гентингтона |
 | |
| Отредактированное микроскопическое изображение среднего шиповидного нейрона (желтый) с телом включения (оранжевый), которое возникает как часть болезненного процесса (ширина изображения 360 мкм ). | |
| Специальность | Неврология |
| Симптомы | Проблемы с моторикой, включая координацию и походку, настроение и умственные способности. [ 1 ] [ 2 ] |
| Осложнения | Пневмония , болезни сердца , травмы в результате падения, самоубийство. [ 3 ] |
| Обычное начало | 30–50 лет [ 4 ] |
| Продолжительность | Долгосрочная перспектива [ 4 ] |
| Причины | Генетический (наследственная или новая мутация) [ 4 ] |
| Метод диагностики | Генетическое тестирование [ 5 ] |
| Дифференциальный диагноз | Хорея Сиденгама , доброкачественная наследственная хорея , волчанка , паранеопластический синдром , болезнь Вильсона [ 6 ] |
| Уход | Поддерживающий уход [ 2 ] |
| Медикамент | Тетрабеназин [ 3 ] |
| Прогноз | 15–20 лет от появления симптомов [ 4 ] |
| Частота | 4–15 из 100 000 (европейское происхождение) [ 1 ] |
| Назван в честь | Джордж Хантингтон |
Болезнь Хантингтона ( БХ ), также известная как хорея Хантингтона , является неизлечимым нейродегенеративным заболеванием. [ 7 ] это в основном наследуется . [ 8 ] Самыми ранними симптомами часто являются едва заметные проблемы с настроением или умственными/психиатрическими способностями. [ 9 ] [ 1 ] Часто за этим следует общее отсутствие координации и неустойчивая походка . [ 2 ] Это также заболевание базальных ганглиев, вызывающее гиперкинетическое двигательное расстройство , известное как хорея . [ 10 ] [ 11 ] По мере прогрессирования заболевания нескоординированные, непроизвольные движения тела при хорее становятся более очевидными. [ 1 ] Физические способности постепенно ухудшаются, пока координация движений не становится затрудненной и человек не может говорить. [ 1 ] [ 2 ] Умственные способности обычно снижаются до деменции , депрессии, апатии и временами импульсивности. [ 9 ] [ 12 ] [ 3 ] Конкретные симптомы несколько различаются у разных людей. [ 1 ] Симптомы обычно начинаются в возрасте от 30 до 50 лет и могут начаться в любом возрасте, но обычно наблюдаются в возрасте около 40 лет. [ 12 ] [ 9 ] [ 3 ] [ 4 ] Заболевание может развиваться раньше в каждом последующем поколении . [ 1 ] Около восьми процентов случаев начинаются в возрасте до 20 лет и известны как ювенильная ГБ , которая обычно проявляется медленными симптомами болезни Паркинсона , а не хореи. [ 3 ]
HD обычно наследуется от больного родителя , который несет мутацию в гене хантингтина ( HTT ). [ 4 ] Однако до 10% случаев обусловлены новой мутацией. [ 1 ] Ген хантингтина предоставляет генетическую информацию о белке хантингтина (Htt). [ 1 ] Расширение CAG-повторов - цитозин аденин - гуанин ( известное как расширение тринуклеотидных повторов ) в гене, кодирующем белок хантингтин, приводит к образованию аномального мутантного белка (mHtt), который постепенно повреждает клетки мозга посредством ряда возможных механизмов. [ 8 ] [ 13 ] Мутантный белок является доминантным , поэтому наличия одного родителя, являющегося носителем этого признака, достаточно, чтобы вызвать заболевание у их детей. Диагноз ставится с помощью генетического тестирования , которое можно провести в любое время, независимо от наличия симптомов. [ 5 ] Этот факт вызывает несколько этических дискуссий: возраст, в котором человек считается достаточно зрелым, чтобы выбрать тестирование; имеют ли родители право на тестирование своих детей; а также обеспечение конфиденциальности и раскрытие результатов испытаний. [ 2 ]
Лекарство от БГ не известно, и на более поздних стадиях требуется постоянный уход. [ 2 ] Лечение может облегчить некоторые симптомы, а в некоторых улучшить качество жизни . [ 3 ] Лучшим доказательством лечения двигательных проблем является тетрабеназин . [ 3 ] HD поражает от 4 до 15 из 100 000 человек европейского происхождения. [ 1 ] [ 3 ] Среди финнов и японцев встречается редко, частота встречаемости в Африке неизвестна. [ 3 ] Заболевание поражает в равной степени мужчин и женщин. [ 3 ] Такие осложнения, как пневмония , болезни сердца и физические травмы в результате падений, сокращают продолжительность жизни; хотя фатальную аспирационную пневмонию обычно называют основной причиной смерти людей с этим заболеванием. [ 14 ] [ 12 ] [ 3 ] Суицид является причиной смерти примерно в 9% случаев. [ 3 ] Смерть обычно наступает через 15–20 лет с момента первого обнаружения заболевания. [ 4 ]
Самое раннее известное описание болезни было сделано в 1841 году американским врачом Чарльзом Оскаром Уотерсом. [ 15 ] Более подробно это состояние описал в 1872 году американский врач Джордж Хантингтон . [ 15 ] Генетическая основа была открыта в 1993 году международным сотрудничеством под руководством Фонда наследственных заболеваний . [ 16 ] [ 17 ] Исследовательские и вспомогательные организации начали формироваться в конце 1960-х годов для повышения осведомленности общественности, оказания поддержки отдельным лицам и их семьям и продвижения исследований. [ 17 ] [ 18 ] Направления исследований включают определение точного механизма заболевания, улучшение моделей животных для помощи в исследованиях, тестирование лекарств и их доставку для лечения симптомов или замедления прогрессирования заболевания, а также изучение таких процедур, как терапия стволовыми клетками с целью замены поврежденные или потерянные нейроны. [ 16 ]
Признаки и симптомы
[ редактировать ]Признаки и симптомы болезни Хантингтона чаще всего становятся заметными в возрасте от 30 до 50 лет, но они могут начаться в любом возрасте. [ 4 ] и проявляется как триада двигательных, когнитивных и психиатрических симптомов. [ 19 ] Если болезнь развивается на ранней стадии, она известна как ювенильная болезнь Хантингтона. [ 20 ] В 50% случаев первыми появляются психиатрические симптомы. [ 19 ] Их прогрессирование часто описывается на ранних стадиях, средних стадиях и поздних стадиях с более ранней продромальной фазой. [ 2 ] На ранних стадиях возникают тонкие изменения личности, проблемы с когнитивными способностями и физическими навыками, раздражительность и перепады настроения, и все это может остаться незамеченным. [ 21 ] [ 22 ] и они обычно предшествуют двигательным симптомам. [ 23 ] Почти у каждого человека с HD в конечном итоге проявляются схожие физические симптомы, но начало, прогрессирование и степень когнитивных и поведенческих симптомов значительно различаются у разных людей. [ 24 ] [ 25 ]
Наиболее характерными начальными физическими симптомами являются судорожные, беспорядочные и неконтролируемые движения, называемые хореей . [ 10 ] Многие люди не осознают своих непроизвольных движений или того, что они им мешают. [ 1 ] Хорея может первоначально проявляться в виде общего беспокойства, небольших непреднамеренно начатых или незавершенных движений, нарушения координации или замедленных саккадических движений глаз . [ 26 ] Эти незначительные двигательные нарушения обычно предшествуют более очевидным признакам двигательной дисфункции как минимум на три года. [ 27 ] явные симптомы, такие как ригидность, корчащиеся движения или ненормальная поза . По мере прогрессирования заболевания появляются [ 26 ] Это признаки того, что поражена система мозга, отвечающая за движение. [ 28 ] Психомоторные функции становятся все более нарушенными, так что затрагиваются любые действия, требующие мышечного контроля. Когда нарушается мышечный контроль, например возникает ригидность или мышечная контрактура, это называется дистонией . Дистония — это неврологическое гиперкинетическое двигательное расстройство, которое приводит к скручиванию или повторяющимся движениям, которые могут напоминать тремор. Распространенными последствиями являются физическая нестабильность, аномальное выражение лица и трудности с жеванием, глотанием и речью . [ 26 ] Нарушения сна и потеря веса также являются сопутствующими симптомами. [ 29 ] Трудности с питанием обычно приводят к потере веса и могут привести к недостаточности питания. [ 30 ] [ 31 ] Потеря веса часто встречается у людей с болезнью Хантингтона и прогрессирует по мере заболевания. Ювенильная БГ обычно прогрессирует быстрее, с более выраженным снижением когнитивных функций, хорея проявляется кратковременно, если вообще проявляется; Вестфальский вариант замедленности движений , ригидности и тремора более типичен для юношеской ГБ, как и судороги . [ 26 ] [ 29 ]
Когнитивные способности прогрессивно ухудшаются и обычно переходят в деменцию . [ 3 ] Особенно страдают исполнительные функции , к которым относятся планирование, когнитивная гибкость, абстрактное мышление , усвоение правил, инициирование соответствующих действий и подавление неподходящих действий. Различные когнитивные нарушения включают трудности с концентрацией внимания на задачах, отсутствие гибкости, отсутствие импульсивности, отсутствие осознания собственного поведения и способностей, а также трудности с обучением или обработкой новой информации. По мере прогрессирования заболевания памяти появляются нарушения . Зарегистрированные нарушения варьируются от дефицита кратковременной памяти до проблем с долговременной памятью , включая дефицит эпизодической (память о своей жизни), процедурной (память тела о том, как выполнять какую-либо деятельность) и рабочей памяти . [ 28 ]
Сообщаемыми нейропсихиатрическими признаками являются тревога , депрессия , снижение проявления эмоций , эгоцентризм , агрессия , компульсивное поведение , галлюцинации и бред . [ 32 ] Другие распространенные психические расстройства могут включать обсессивно-компульсивное расстройство , манию , бессонницу и биполярное расстройство . Также наблюдались трудности с распознаванием негативных высказываний других людей. [ 28 ] Распространенность составляет этих симптомов сильно различается в разных исследованиях: по оценкам, распространенность психических расстройств в течение жизни от 33 до 76%. [ 32 ] Для многих больных и членов их семей эти симптомы являются одними из наиболее тревожных аспектов заболевания, часто влияя на повседневное функционирование и являясь причиной помещения в специальные учреждения . [ 32 ] Ранние поведенческие изменения при ГБ приводят к повышенному риску самоубийства. [ 10 ] Часто у людей снижается осознание хореи, когнитивных и эмоциональных нарушений. [ 33 ]
Мутантный хантингтин экспрессируется по всему организму и связан с аномалиями в периферических тканях, которые непосредственно вызваны такой экспрессией за пределами мозга. Эти нарушения включают мышечную атрофию , сердечную недостаточность , нарушение толерантности к глюкозе , потерю веса , остеопороз и атрофию яичек . [ 34 ]
Генетика
[ редактировать ]У каждого есть две копии гена хантингтина ( HTT ), который кодирует белок хантингтина (Htt). HTT также называют геном HD и геном IT15 (интересный транскрипт 15). Часть этого гена представляет собой повторяющийся участок, называемый расширением тринуклеотидных повторов – короткий повтор , длина которого варьируется у разных людей и может меняться в зависимости от поколения. Если повтор присутствует в здоровом гене, динамическая мутация может увеличить количество повторов и привести к дефектному гену. Когда длина этого повторяющегося участка достигает определенного порога, он производит измененную форму белка, называемую мутантным белком хантингтина (mHtt). Различные функции этих белков являются причиной патологических изменений, которые, в свою очередь, вызывают симптомы заболевания. Мутация болезни Хантингтона является генетически доминантной и почти полностью пенетрантной ; человека Мутация любого из аллелей HTT вызывает заболевание. Наследуется не по полу, а по длине повторяющегося участка гена; следовательно, на тяжесть заболевания может влиять пол больного родителя. [ 26 ]
Генетическая мутация
[ редактировать ]HD является одним из нескольких нарушений тринуклеотидных повторов , которые вызваны длиной повторяющегося участка гена, превышающей нормальный диапазон. [ 26 ] Ген HTT расположен на коротком плече . 4-й хромосомы [ 26 ] в 4p16.3. HTT содержит последовательность трех оснований ДНК — цитозин-аденин-гуанин (CAG), повторяющуюся несколько раз (т. е.… CAGCAGCAG…), известную как тринуклеотидный повтор. [ 26 ] CAG — это трехбуквенный генетический код ( кодон ) аминокислоты глутамин , поэтому серия из них приводит к образованию цепи глютамина, известной как полиглутаминовый тракт (или полиQ-тракт), и повторяющейся части гена, область полиQ . [ 35 ]
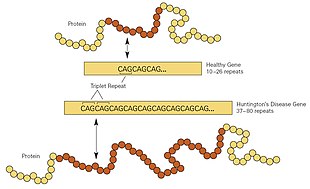
| Повторить счет | Классификация | Статус заболевания | Риск для потомства |
|---|---|---|---|
| <27 | Нормальный | Не будет затронуто | Никто |
| 27–35 | Средний | Не будет затронуто | Повышен, но <50% |
| 36–39 | Пониженная пенетрантность | Может быть затронуто или не затронуто | 50% |
| 40+ | Полная пенетрантность | Будет затронуто | 50% |
Как правило, у людей имеется менее 36 повторяющихся глютаминов в области PolyQ, что приводит к выработке цитоплазматического белка хантингтина. [ 26 ] Однако последовательность из 36 или более глютаминов приводит к образованию белка с другими характеристиками. [ 26 ] Эта измененная форма, называемая мутантным хантингтином (mHtt), увеличивает скорость распада определенных типов нейронов . Области мозга имеют различное количество и зависимость от этих типов нейронов и подвергаются соответствующему воздействию. [ 26 ] Как правило, количество CAG-повторов зависит от степени поражения этого процесса и составляет около 60% вариаций возраста появления симптомов. Остальные вариации связаны с окружающей средой и другими генами, которые модифицируют механизм БГ. [ 26 ] Примерно от 36 до 39 повторов приводят к форме заболевания со сниженной пенетрантностью, с гораздо более поздним началом и более медленным прогрессированием симптомов. В некоторых случаях начало может быть настолько поздним, что симптомы вообще не замечаются. [ 26 ] При очень большом количестве повторов (более 60) начало БГ может произойти в возрасте до 20 лет, что известно как ювенильная БГ. Ювенильный БГ обычно относится к вестфальскому варианту, который характеризуется замедленностью движений, ригидностью и тремором. Это составляет около 7% носителей HD. [ 36 ] [ 37 ]
Наследование
[ редактировать ]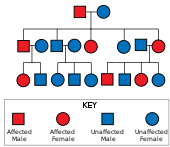
Болезнь Хантингтона имеет аутосомно-доминантное наследование, что означает, что больной человек обычно наследует одну копию гена с расширенным тринуклеотидным повтором (мутантный аллель ) от больного родителя. [ 26 ] Поскольку пенетрантность мутации очень высока, заболеванием заболеют те, у кого есть мутированная копия гена. При этом типе наследования каждый потомок больного человека имеет 50% риск унаследовать мутантный аллель, поэтому у него возникает заболевание (см. рисунок). Эта вероятность не зависит от пола. [ 38 ] Гены, зависящие от пола или сцепленные с полом, — это признаки, которые обнаруживаются в X или Y-хромосомах. [ 39 ]
Тринуклеотидные повторы CAG, насчитывающие более 28, нестабильны во время репликации , и эта нестабильность увеличивается с увеличением количества присутствующих повторов. [ 26 ] Обычно это приводит к новым расширениям по мере прохождения поколений ( динамические мутации ) вместо воспроизведения точной копии тринуклеотидного повтора. [ 26 ] Это приводит к изменению количества повторов в последующих поколениях, так что незатронутый родитель с «промежуточным» количеством повторов (28–35) или «пониженной пенетрантностью» (36–40) может передать копию гена. с увеличением количества повторов, что приводит к полностью проникающему ГД. [ 26 ] Более ранний возраст начала и большая тяжесть заболевания в последующих поколениях из-за увеличения числа повторов известны как генетическое предвосхищение . [ 1 ] Нестабильность выше в сперматогенезе , чем в оогенезе ; [ 26 ] аллели, унаследованные по материнской линии, обычно имеют одинаковую длину повторов, тогда как аллели, унаследованные по отцовской линии, имеют более высокий шанс увеличения длины. [ 26 ] [ 40 ] Редко болезнь Хантингтона вызывается новой мутацией , когда ни один из родителей не имеет более 36 CAG-повторов. [ 41 ]
В редких ситуациях, когда оба родителя имеют расширенный ген HD, риск увеличивается до 75%, а когда у одного из родителей есть две расширенные копии, риск составляет 100% (пострадают все дети). Лица, у которых поражены оба гена, встречаются редко. Некоторое время считалось, что БГ является единственным заболеванием, при котором наличие второго мутировавшего гена не влияет на симптомы и прогрессирование. [ 42 ] но с тех пор было обнаружено, что оно может влиять на фенотип и скорость прогрессирования. [ 26 ] [ 43 ]
Механизмы
[ редактировать ]Белок хантингтин взаимодействует с более чем 100 другими белками и, по-видимому, выполняет множество функций. [ 44 ] Поведение мутировавшего белка (mHtt) до конца не изучено, но он токсичен для определенных типов клеток, особенно клеток головного мозга . Раннее поражение наиболее очевидно в подкорковых базальных ганглиях , первоначально в полосатом теле , но по мере прогрессирования заболевания поражаются и другие области головного мозга, включая области коры головного мозга . Ранние симптомы обусловлены функциями полосатого тела и его корковых связей, а именно контролем над движением, настроением и высшими когнитивными функциями. [ 26 ] Метилирование ДНК также, по-видимому, изменяется при БГ. [ 45 ]
Функция Хантингтина
[ редактировать ]Htt экспрессируется во всех клетках, при этом самые высокие концентрации обнаруживаются в мозге и семенниках , а умеренные количества — в печени , сердце и легких . Его функции неясны, но он взаимодействует с белками, участвующими в транскрипции , передаче сигналов в клетках и внутриклеточном транспорте . [ 46 ] У животных, генетически модифицированных для проявления HD, было идентифицировано несколько функций Htt. [ 47 ] У этих животных Htt важен для эмбрионального развития, поскольку его отсутствие связано с эмбриональной гибелью. Считается, что каспаза , фермент, который играет роль в катализе апоптоза , активируется мутировавшим геном путем повреждения системы убиквитин-протеаза. Он также действует как антиапоптотический агент, предотвращая запрограммированную гибель клеток , и контролирует выработку мозгового нейротрофического фактора — белка, который защищает нейроны и регулирует их создание во время нейрогенеза . Htt также облегчает синаптический везикулярный транспорт и синаптическую передачу и контролирует транскрипцию нейрональных генов. [ 47 ] Если экспрессия Htt увеличивается, выживаемость клеток головного мозга улучшается, а эффекты mHtt уменьшаются, тогда как когда экспрессия Htt снижается, результирующие характеристики в большей степени соответствуют наблюдаемым в присутствии mHtt. [ 47 ] Соответственно, считается, что заболевание вызвано не неадекватным производством Htt, а токсическим усилением функции mHtt в организме. [ 26 ]
Клеточные изменения
[ редактировать ]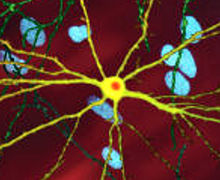
Токсическое действие mHtt может проявляться и вызывать патологию ГБ через множественные клеточные изменения. [ 48 ] [ 49 ] В своей мутантной (расширенной полиглутамином) форме белок более склонен к расщеплению, в результате чего образуются более короткие фрагменты, содержащие расширение полиглутамина. [ 48 ] Эти белковые фрагменты склонны к неправильному сворачиванию и агрегации, образуя фибриллярные агрегаты, в которых ненативные полиглутаминовые β-цепи из нескольких белков связаны друг с другом водородными связями. [ 13 ] Эти агрегаты имеют ту же фундаментальную архитектуру перекрестного бета- амилоида , что и при других заболеваниях отложения белка . [ 50 ] Со временем агрегаты накапливаются, образуя тела включения внутри клеток, что в конечном итоге нарушает функцию нейронов. [ 13 ] [ 48 ] Тельца включения были обнаружены как в ядре клетки , так и в цитоплазме . [ 48 ] Тельца включения в клетках головного мозга являются одним из самых ранних патологических изменений, и некоторые эксперименты показали, что они могут быть токсичными для клетки, но другие эксперименты показали, что они могут формироваться как часть защитного механизма организма и помогать защищать клетки. [ 48 ]
Было идентифицировано несколько путей, с помощью которых mHtt может вызывать гибель клеток. К ним относятся воздействие на белки-шапероны , которые помогают сворачивать белки и удалять неправильно свернутые; взаимодействие с каспазами , которые играют роль в процессе удаления клеток ; токсическое воздействие глютамина на нервные клетки ; нарушение производства энергии внутри клеток; и влияние на экспрессию генов. [ 13 ] [ 51 ]
Было обнаружено, что мутантный белок хантингтин играет ключевую роль в дисфункции митохондрий . [ 46 ] Нарушение митохондриального транспорта электронов может привести к более высокому уровню окислительного стресса и выделению активных форм кислорода . [ 52 ]
Известно, что глутамин эксайтотоксичен в больших количествах и может вызвать повреждение многочисленных клеточных структур. Избыток глутамина не обнаруживается при БГ, но взаимодействия измененного белка хантингтина с многочисленными белками нейронов приводят к повышенной уязвимости к глютамину. Считается, что повышенная уязвимость приводит к эксайтотоксическим эффектам из-за нормального уровня глютамина. [ 13 ]
Макроскопические изменения
[ редактировать ]
Первоначально повреждение головного мозга является регионально специфичным: дорсальное полосатое тело в подкорковых базальных ганглиях в первую очередь поражается , а затем вовлекается кора во всех областях. [ 53 ] [ 54 ] Другие пораженные области базальных ганглиев включают черную субстанцию ; вовлечение коры включает корковые слои 3, 5 и 6 ; также очевидно вовлечение гиппокампа , клеток Пуркинье в мозжечке , латеральных туберальных ядер гипоталамуса и частей таламуса . [ 26 ] Эти области поражаются в зависимости от их структуры и типов содержащихся в них нейронов, уменьшаясь в размерах по мере потери клеток. [ 26 ] стриарного тела шипиковые нейроны Наиболее уязвимыми являются , особенно те, которые проецируются в сторону наружного бледного шара , при этом вставочные нейроны и шипистые клетки, выступающие во внутренний бледный шар, поражаются меньше. [ 26 ] [ 55 ] вызывает аномальное увеличение астроцитов HD также и активацию иммунных клеток мозга, микроглии . [ 56 ]
Базальные ганглии играют ключевую роль в контроле движений и поведения. Их функции до конца не изучены, но теории предполагают, что они являются частью когнитивно- исполнительной системы. [ 28 ] и цепь двигателя. [ 57 ] Базальные ганглии обычно подавляют большое количество цепей, генерирующих определенные движения. Чтобы инициировать определенное движение, кора головного мозга посылает сигнал базальным ганглиям, который вызывает прекращение торможения. Повреждение базальных ганглиев может привести к тому, что высвобождение или восстановление торможения будет беспорядочным и неконтролируемым, что приводит к неловкому началу движения или к непреднамеренному инициированию движений или к остановке движения до или после его запланированного завершения. Накопление повреждений в этой области вызывает характерные беспорядочные движения, связанные с ГБ, известные как хорея, дискинезия . [ 57 ] Из-за неспособности базальных ганглиев подавлять движения у людей, затронутых этим заболеванием, неизбежно снижается способность произносить речь и глотать пищу и жидкости (дисфагия). [ 58 ]
Транскрипционная дисрегуляция
[ редактировать ]CREB-связывающий белок (CBP), корегулятор транскрипции, необходим для функционирования клеток, поскольку в качестве коактиватора значительного числа промоторов он активирует транскрипцию генов, участвующих в путях выживания. [ 51 ] CBP содержит домен ацетилтрансферазы , с которым HTT связывается через свой полиглутамин-содержащий домен. [ 59 ] При аутопсии мозга тех, кто страдал болезнью Хантингтона, также было обнаружено невероятное снижение количества CBP. [ 60 ] Кроме того, когда CBP сверхэкспрессируется, смертность, вызванная полиглутамином, уменьшается, что еще раз демонстрирует, что CBP играет важную роль в болезни Хантингтона и нейронах в целом. [ 51 ]
Диагностика
[ редактировать ]Диагноз начала ГБ можно поставить после появления физических симптомов, характерных для заболевания. [ 26 ] Генетическое тестирование может быть использовано для подтверждения физического диагноза, если в семейном анамнезе не существует БГ. Еще до появления симптомов генетическое тестирование может подтвердить, несет ли человек или эмбрион расширенную копию тринуклеотидного повтора (CAG) в гене HTT , вызывающем заболевание. Доступны генетические консультации , которые предоставят советы и рекомендации на протяжении всей процедуры тестирования, а также о последствиях подтвержденного диагноза. Эти последствия включают влияние на психологию человека, карьеру, решения по планированию семьи, родственников и отношения. Несмотря на доступность пресимптоматического тестирования, только 5% из тех, кто подвержен риску наследования БГ, решают это сделать. [ 26 ]
Клинический
[ редактировать ]
Физикальное обследование , иногда в сочетании с психологическим обследованием , позволяет определить, началось ли начало заболевания. [ 26 ] Чрезмерные непреднамеренные движения какой-либо части тела часто являются поводом для обращения к врачу. Если они происходят внезапно и имеют случайное время и распределение, они предполагают диагноз ГБ. Когнитивные или поведенческие симптомы редко являются первыми диагностируемыми симптомами; их обычно распознают только задним числом или по мере дальнейшего развития. Насколько далеко продвинулось заболевание, можно измерить с помощью единой шкалы оценки болезни Хантингтона, которая представляет собой общую систему оценок, основанную на двигательных, поведенческих, когнитивных и функциональных оценках. [ 62 ] [ 63 ] Медицинские методы визуализации , такие как КТ или МРТ , могут выявить атрофию хвостатых ядер на ранних стадиях заболевания, как показано на рисунке справа, но эти изменения сами по себе не являются диагностическими признаками БГ. Церебральная атрофия может наблюдаться на поздних стадиях заболевания. Функциональные методы нейровизуализации , такие как функциональная магнитно-резонансная томография (фМРТ) и позитронно-эмиссионная томография (ПЭТ), могут показать изменения в активности мозга до появления физических симптомов, но они являются экспериментальными инструментами и не используются клинически. [ 26 ]
Прогнозирующее генетическое тестирование
[ редактировать ]Поскольку БГ наследуется по аутосомно-доминантному типу, у людей, которые подвержены риску ее унаследования, существует сильная мотивация обратиться за диагнозом. Генетический тест на БГ состоит из анализа крови, в ходе которого подсчитывают количество повторов CAG в каждом из аллелей HTT . [ 64 ] Отсечки даны следующим образом:
- При 40 и более повторах CAG существует аллель полной пенетрантности (FPA). [ 65 ] « положительный тест К этому случаю обычно относятся » или «положительный результат». Положительный результат не считается диагнозом, поскольку его можно получить за десятилетия до появления симптомов. Однако отрицательный результат теста означает, что человек не является носителем расширенной копии гена и у него не разовьется HD. [ 26 ] Тест сообщит человеку, у которого изначально была 50% вероятность унаследовать заболевание, увеличится ли риск до 100% или будет устранен. У людей с положительным результатом теста на заболевание БГ когда-нибудь в течение жизни разовьется, при условии, что они проживут достаточно долго, чтобы появилось заболевание. [ 26 ]
- При количестве повторов от 36 до 39 аллель неполной или пониженной пенетрантности (RPA) может вызывать симптомы, обычно в более позднем взрослом возрасте. [ 65 ] Максимальный риск того, что у человека с РПА проявятся симптомы в 65 лет, составляет 60%, а в 75 лет — 70%. [ 65 ]
- При 27–35 повторах промежуточный аллель (IA) или большой нормальный аллель не связан с симптоматическим заболеванием у тестируемого человека, но может расширяться при дальнейшем наследовании, вызывая симптомы у потомства. [ 65 ]
- При 26 и менее повторах результат не связан с БГ. [ 65 ]
Тестирование до появления симптомов — это событие, которое меняет жизнь, и это очень личное решение. [ 26 ] Основная причина выбора теста на HD – это помощь в принятии решений о карьере и семье. [ 26 ] Прогностическое тестирование на болезнь Хантингтона доступно с помощью анализа сцепления (который требует тестирования нескольких членов семьи) с 1986 года и с помощью прямого анализа мутаций с 1993 года. [ 66 ] В то время опросы показали, что 50–70% лиц из группы риска были бы заинтересованы в прохождении тестирования, но поскольку предлагалось прогностическое тестирование, гораздо меньше людей решили пройти тестирование. [ 67 ] Более 95% людей с риском наследования ГБ не проходят тестирование, главным образом потому, что оно не лечится. [ 26 ] Ключевой проблемой является беспокойство, которое человек испытывает из-за незнания, разовьется ли у него в конечном итоге ГБ, по сравнению с влиянием положительного результата. [ 26 ] Независимо от результата, уровень стресса снижается через два года после тестирования, но риск самоубийства увеличивается после положительного результата теста. [ 26 ] Лица, у которых установлено, что это расстройство не унаследовано, могут испытывать чувство вины выжившего перед затронутыми членами семьи. [ 26 ] Другие факторы, принимаемые во внимание при рассмотрении вопроса о тестировании, включают возможность дискриминации и последствия положительного результата, что обычно означает, что у родителя есть пораженный ген и что братья и сестры человека будут подвергаться риску его унаследования. [ 26 ] В одном исследовании генетическая дискриминация была обнаружена у 46% людей, подверженных риску болезни Хантингтона. Это происходило чаще в личных отношениях, чем в сфере медицинского страхования или трудовых отношениях. [ 68 ] Генетическое консультирование при БГ может предоставить информацию, советы и поддержку для принятия первоначальных решений, а затем, если это будет выбрано, на всех этапах процесса тестирования. [ 69 ] Из-за последствий этого теста пациенты, желающие пройти тестирование, должны пройти три консультации, на которых предоставят информацию о болезни Хантингтона. [ 70 ]
Консультации и рекомендации по использованию генетического тестирования на БГ стали моделями для других генетических нарушений, таких как аутосомно-доминантная мозжечковая атаксия . [ 26 ] [ 71 ] [ 72 ] Предсимптоматическое тестирование на БГ также повлияло на тестирование на другие заболевания с генетическими вариантами, такие как поликистоз почек , семейная болезнь Альцгеймера и рак молочной железы . [ 71 ] Европейская сеть качества молекулярной генетики ежегодно публикует схему внешней оценки качества молекулярно-генетического тестирования на это заболевание и разработала рекомендации по передовой практике генетического тестирования на БГ, чтобы помочь в тестировании и представлении результатов. [ 73 ]
Преимплантационная генетическая диагностика
[ редактировать ]Эмбрионы , полученные с помощью экстракорпорального оплодотворения, могут быть генетически протестированы на БГ с использованием преимплантационной генетической диагностики . Этот метод, при котором одна или две клетки извлекаются из эмбриона, состоящего обычно из 4–8 клеток, а затем проверяются на наличие генетических аномалий, может затем использоваться для обеспечения того, чтобы эмбрионы, пораженные генами HD, не были имплантированы, поэтому ни один потомок не унаследует болезнь. Некоторые формы преимплантационной генетической диагностики – тестирование на неразглашение или тестирование на исключение – позволяют людям из группы риска иметь потомство без БГ, не раскрывая свой собственный родительский генотип, не давая информации о том, предначертано ли им самим развить БГ. При тестировании на исключение ДНК эмбриона сравнивают с ДНК родителей, бабушек и дедушек, чтобы избежать наследования хромосомной области, содержащей ген HD, от больного дедушки и бабушки. При тестировании с сохранением конфиденциальности в матке заменяются только здоровые эмбрионы, при этом родительский генотип и, следовательно, родительский риск развития БГ никогда не раскрываются. [ 74 ] [ 75 ]
Пренатальное тестирование
[ редактировать ]Пренатальная диагностика эмбриона или плода в утробе матери также возможна с использованием генетического материала плода, полученного при заборе ворсин хориона . Амниоцентез можно провести , если беременность продолжается, в течение 14–18 недель. В ходе этой процедуры амниотическая жидкость, окружающая ребенка, исследуется на наличие признаков мутации HD. [ 76 ] Это также можно сочетать с тестированием на исключение, чтобы избежать раскрытия родительского генотипа. Пренатальное тестирование можно проводить, если у родителей был диагностирован БГ, когда у них было генетическое тестирование, показывающее экспансию гена HTT , или когда у них есть 50% вероятность унаследовать заболевание. Родители могут быть проконсультированы о возможных вариантах, включая прерывание беременности , а также о трудностях ребенка с выявленным геном. [ 77 ] [ 78 ]
Кроме того, при беременности из группы риска из-за пораженного партнера-мужчины неинвазивная пренатальная диагностика может быть выполнена путем анализа бесклеточной ДНК плода в образце крови, взятом у матери (через венепункцию ) между 6 и 12 неделями беременности. [ 65 ] Риск выкидыша, связанный с процедурой, отсутствует. [ 65 ]
Дифференциальный диагноз
[ редактировать ]Около 99% диагнозов БГ, основанных на типичных симптомах и семейном анамнезе заболевания, подтверждаются генетическим тестированием на наличие расширенного тринуклеотидного повтора, вызывающего БГ. Большинство из оставшихся называются HDL-подобными (HDL) синдромами . [ 26 ] [ 79 ] Причина большинства заболеваний ЛПВП неизвестна, но те, причины которых известны, обусловлены мутациями в гене прионного белка (ЛПВП1), гене юнктофилина 3 (ЛПВП2), рецессивно наследуемом неизвестном гене (ЛПВП3 – обнаружен только в двух семьях и плохо изучен). понятен) и ген, кодирующий белок, связывающий ТАТА-бокс ( SCA17, иногда называемый HDL4 ). Другими аутосомно-доминантными заболеваниями, которые могут быть ошибочно приняты за БГ, являются дентаторубрально-паллидолуизова атрофия и нейроферритинопатия . Кроме того, некоторые аутосомно-рецессивные заболевания напоминают спорадические случаи БГ. К ним относятся хореальный акантоцитоз и нейродегенерация, связанная с пантотенаткиназой . Одним из Х-сцепленных заболеваний этого типа является синдром МакЛеода . [ 79 ]
Управление
[ редактировать ]
Доступны методы лечения, позволяющие уменьшить тяжесть некоторых симптомов ГБ. [ 80 ] Для многих из этих методов лечения доказательства, подтверждающие их эффективность в лечении симптомов ГБ, являются неполными. [ 26 ] [ 81 ] По мере прогрессирования заболевания способность заботиться о себе снижается, и тщательно организованный междисциплинарный уход становится все более необходимым. [ 26 ] Хотя относительно небольшое количество исследований упражнений и методов лечения показали эффективность реабилитации когнитивных симптомов ГБ, некоторые данные показывают полезность физиотерапии , трудотерапии и логопеда . [ 26 ]
Терапия
[ редактировать ]Потеря веса и проблемы с питанием из-за дисфагии и других нарушений координации мышц являются обычным явлением, что делает контроль питания все более важным по мере прогрессирования заболевания. [ 26 ] В жидкости можно добавлять загустители , поскольку более густые жидкости легче и безопаснее глотать. [ 26 ] Напоминание пострадавшему о необходимости есть медленно и брать в рот более мелкие кусочки пищи также может быть полезно для предотвращения удушья. [ 26 ] Если прием пищи становится слишком опасным или неудобным, можно использовать чрескожную эндоскопическую гастростомию . Эта трубка для кормления, постоянно прикрепленная через брюшную полость к желудку , снижает риск аспирации пищи и обеспечивает лучший контроль питания. [ 82 ] Рекомендуется обследование и лечение у логопедов с опытом работы с болезнью Гентингтона. [ 26 ]
Люди с болезнью Хантингтона могут обратиться к физиотерапевту, чтобы узнать о неинвазивных и немедикаментозных способах лечения физических симптомов. Физиотерапевты могут проводить оценку и предотвращение риска падения, а также выполнять упражнения на укрепление, растяжку и сердечно-сосудистые упражнения. средства для ходьбы При необходимости могут быть назначены . Физиотерапевты также назначают дыхательные упражнения и техники очистки дыхательных путей при развитии респираторных проблем. [ 83 ] Согласованные рекомендации по физиотерапии при болезни Хантингтона были разработаны Европейской сетью HD. [ 83 ] Целью раннего реабилитационного вмешательства является предотвращение потери функции. Участие в программах реабилитации на ранней и средней стадии заболевания может быть полезным, поскольку приводит к долгосрочному поддержанию двигательных и функциональных показателей. Реабилитация на поздней стадии направлена на компенсацию двигательных и функциональных потерь. [ 84 ] Для долгосрочного независимого лечения терапевт может разработать программы домашних упражнений для подходящих людей. [ 85 ]
Кроме того, все большее число людей с ГБ обращаются к паллиативной помощи , целью которой является улучшение качества жизни посредством лечения симптомов и стресса серьезного заболевания в дополнение к другим методам лечения. [ 86 ]
Лекарства
[ редактировать ]
Тетрабеназин был одобрен в 2000 году для лечения хореи при болезни Хантингтона в ЕС и в 2008 году в США. [ 87 ] Хотя другие препараты использовались « не по назначению », тетрабеназин был первым одобренным средством лечения болезни Хантингтона в США. Это соединение известно с 1950-х годов. Альтернативой тетрабеназину является амантадин, однако данные о его безопасности и эффективности ограничены. [ 88 ]
Другие препараты, которые помогают уменьшить хорею, включают нейролептики и бензодиазепины . [ 22 ] Гипокинезию и ригидность, особенно в юношеских случаях, можно лечить противопаркинсоническими препаратами, а миоклоническую гиперкинезию — вальпроевой кислотой . [ 22 ] Предварительные данные показали, что этилэйкозапентаеновая кислота улучшает двигательные симптомы через год. [ 89 ] В 2017 году дейтетрабеназин , более тяжелую форму препарата тетрабеназина для лечения хореи при ГБ. FDA одобрило [ 90 ] Он продается как Аустедо.
Психиатрические симптомы можно лечить с помощью лекарств, аналогичных тем, которые используются среди населения в целом. [ 26 ] [ 81 ] Селективные ингибиторы обратного захвата серотонина и миртазапин рекомендуются при депрессии, а атипичные антипсихотики — при психозе и поведенческих проблемах. [ 81 ] Рекомендуется участие специалиста нейропсихиатра, поскольку людям может потребоваться длительное лечение с применением нескольких препаратов в комбинации. [ 26 ]
Растительные лекарства
[ редактировать ]медицине экспериментировали с рядом альтернативных методов лечения В аюрведической с использованием продуктов растительного происхождения, хотя ни один из них не дал убедительных доказательств эффективности. Недавнее исследование показало, что стромальная процессинговая пептидаза (SPP), синтетический фермент, обнаруженный в хлоропластах растений , предотвращает агрегацию белков, связанных с болезнью Хантингтона. [ 91 ] Однако для подтверждения его истинного терапевтического потенциала необходимы повторные исследования и клиническая проверка.
Образование
[ редактировать ]Семьи людей и общество в целом, унаследовавшие БГ или находящиеся под угрозой ее наследования, имеют опыт БГ в течение нескольких поколений, но могут не знать о недавних прорывах в понимании этого заболевания и о доступности генетического тестирования. Генетическое консультирование приносит пользу этим людям, обновляя их знания, стремясь развеять любые необоснованные убеждения, которые у них могут быть, и помогая им обдумать свои будущие варианты и планы. Программа обучения пациентов по поводу болезни Хантингтона была создана, чтобы помочь в обучении членов семьи, лиц, осуществляющих уход, и тех, у кого диагностирована болезнь Хантингтона. [ 92 ] Также рассматривается информация, касающаяся выбора планирования семьи, организации ухода и других соображений. [ 26 ] [ 93 ]
Прогноз
[ редактировать ]На длину тринуклеотидного повтора приходится 60% вариаций возраста появления симптомов и скорости их прогрессирования. Более длительное повторение приводит к более раннему началу заболевания и более быстрому прогрессированию симптомов. [ 26 ] [ 94 ] У людей с более чем шестьюдесятью повторами заболевание часто развивается в возрасте до 20 лет, тогда как у людей с менее чем 40 повторами заболевание может оставаться бессимптомным. [ 95 ] Остальные вариации обусловлены факторами окружающей среды и другими генами, влияющими на механизм заболевания. [ 26 ]
Ожидаемая продолжительность жизни при БГ обычно составляет от 10 до 30 лет после появления видимых симптомов. [ 26 ] Ожидаемая продолжительность жизни при ювенильной болезни Хантингтона составляет 10 лет после появления видимых симптомов. Большинство опасных для жизни осложнений возникают в результате мышечной координации и, в меньшей степени, поведенческих изменений, вызванных снижением когнитивных функций. Наибольший риск представляет собой пневмония , которая приводит к смерти одной трети пациентов с ГБ. Поскольку способность синхронизировать движения ухудшается, трудности с очисткой легких и повышенный риск аспирации еды или питья увеличивают риск заражения пневмонией . Вторым по величине риском являются болезни сердца , которые являются причиной почти четверти смертей среди людей с ГБ. [ 96 ] Самоубийство является третьей по значимости причиной смертности: 7,3% больных ГБ покончили с собой и до 27% пытались это сделать. Неясно, в какой степени на суицидальные мысли влияют поведенческие симптомы, поскольку они означают желание избежать более поздних стадий заболевания. [ 97 ] [ 98 ] [ 99 ] Суицид представляет собой наибольший риск этого заболевания до постановки диагноза и на средних стадиях развития на протяжении всего заболевания. Другие связанные с этим риски включают удушье; из-за неспособности глотать, физических травм в результате падений и недоедания. [ 96 ] [ 20 ]
Эпидемиология
[ редактировать ]Позднее начало болезни Хантингтона означает, что она обычно не влияет на репродуктивную функцию. [ 26 ] в мире составляет 5–10 случаев на 100 000 человек. Распространенность ГБ [ 100 ] [ 101 ] но сильно различается географически в зависимости от этнической принадлежности, местной миграции и прошлых моделей иммиграции. [ 26 ] Распространенность одинакова для мужчин и женщин. Уровень заболеваемости самый высокий у людей западноевропейского происхождения, составляя в среднем около семи на 100 000 человек, и ниже в остальном мире; например, один на миллион человек азиатского и африканского происхождения. Эпидемиологическое исследование распространенности болезни Хантингтона в Великобритании в период с 1990 по 2010 год, проведенное в 2013 году, показало, что средний показатель распространенности в Великобритании составил 12,3 на 100 000 человек. [ 26 ] [ 102 ] Кроме того, в некоторых локализованных районах распространенность гораздо выше, чем в среднем по региону. [ 26 ] Один из самых высоких показателей заболеваемости наблюдается в изолированных популяциях региона озера Маракайбо в Венесуэле , где ГБ поражает до 700 на 100 000 человек. [ 26 ] [ 103 ] Другие области высокой локализации были обнаружены на Тасмании и в отдельных регионах Шотландии , Уэльса и Швеции . [ 99 ] Увеличение распространенности в некоторых случаях происходит из-за эффекта местного основателя – исторической миграции носителей в район географической изоляции . [ 99 ] [ 104 ] Некоторые из этих носителей были обнаружены на протяжении сотен лет с помощью генеалогических исследований. [ 99 ] Генетические гаплотипы также могут дать представление о географических вариациях распространенности. [ 99 ] [ 105 ] В Исландии , напротив, распространенность довольно низкая — 1 на 100 000, несмотря на то, что исландцы как народ происходят от ранних германских племен Скандинавии, которые также дали начало шведам ; все случаи, за исключением одного, произошедшего почти два столетия назад, произошли от потомков пары, жившей в начале 19 века. [ 106 ] В Финляндии также низкий уровень заболеваемости – всего 2,2 на 100 000 человек. [ 107 ]
До открытия генетического теста статистика могла включать только клинический диагноз, основанный на физических симптомах и семейном анамнезе БГ, исключая тех, кто умер от других причин до постановки диагноза. Эти случаи теперь могут быть включены в статистику; и по мере того, как тест становится более доступным, оценки распространенности и заболеваемости расстройством, вероятно, возрастут. [ 99 ] [ 108 ]
История
[ редактировать ]
В прошлые века различные виды хореи иногда назывались такими именами, как танец Святого Витта , практически без понимания их причины или типа в каждом случае.
Первое определенное упоминание о ГБ было в письме Чарльза Оскара Уотерса (1816–1892), опубликованном в первом издании « Данглисона Робли Практики медицины» в 1842 году. [ 110 ] Уотерс описал «форму хореи, в просторечии называемую магрумами», включая точные описания хореи, ее прогрессирования и сильной наследственности заболевания. [ 111 ] В 1846 году Чарльз Роллин Горман (1817–1879) заметил, что более высокая распространенность, по-видимому, наблюдалась в локализованных регионах. [ 112 ] [ 111 ] Независимо от Гормана и Уотерса, студентов Данглисона в Медицинском колледже Джефферсона в Филадельфии, [ 113 ] Йохан Кристиан Лунд (1830–1906) также произвел раннее описание в 1860 году. [ 111 ] Он особо отметил, что в Сетесдалене , уединенной горной долине в Норвегии , высокая распространенность деменции была связана с паттерном подергивания двигательных расстройств, который передавался в семьях. [ 114 ]
Первое подробное описание болезни было сделано Джорджем Хантингтоном в 1872 году. Изучая совокупную историю болезни нескольких поколений семей, демонстрирующих схожие симптомы, он понял, что их состояния должны быть связаны между собой; в своей первой статье он представил подробное и точное определение болезни. Хантингтон описал точную картину наследования аутосомно-доминантного заболевания за несколько лет до повторного открытия учеными менделевского наследования .
О своей наследственной природе. Когда у одного или обоих родителей наблюдаются проявления болезни... один или несколько потомков почти всегда страдают от этой болезни... Но если случайно эти дети проживут жизнь без нее, нить оборвется и внуки и правнуки первых шейкеров могут быть уверены, что они свободны от этой болезни. [ 109 ] [ 115 ]
Сэр Уильям Ослер интересовался этим расстройством и хореей в целом и был впечатлен статьей Хантингтона, в которой говорилось: «В истории медицины есть несколько случаев, когда болезнь была описана более точно, более наглядно или более кратко». [ 116 ] [ 111 ] [ 117 ] Постоянный интерес Ослера к ГБ в сочетании с его влиянием в области медицины помог быстро распространить осведомленность и знания об этом заболевании среди медицинского сообщества. [ 111 ] Большой интерес проявили учёные Европы, в том числе Луи Теофиль Жозеф Ландузи , Дезире-Маглуар Бурневиль , Камилло Гольджи и Жозеф Жюль Дежерин , и до конца века большая часть исследований ГБ имела европейское происхождение. [ 111 ] К концу XIX века исследования и отчеты о БГ были опубликованы во многих странах, и это заболевание было признано заболеванием во всем мире. [ 111 ]
Во время повторного открытия менделевского наследования на рубеже 20-го века БГ экспериментально использовалась как пример аутосомно-доминантного наследования. [ 111 ] Английский биолог Уильям Бейтсон использовал родословные больных семей, чтобы установить, что HD имеет аутосомно-доминантный тип наследования. [ 118 ] [ 113 ] Сильный характер наследования побудил нескольких исследователей, в том числе Смита Эли Джеллиффа , попытаться отследить и связать членов семьи, участвовавших в предыдущих исследованиях. [ 111 ] Джеллифф собрал информацию со всего Нью-Йорка и опубликовал несколько статей, касающихся генеалогии ГБ в Новой Англии . [ 119 ] Исследование Джеллиффа вызвало интерес у его друга по колледжу Чарльза Давенпорта , который поручил Элизабет Манси провести первое полевое исследование семей с HD на восточном побережье Соединенных Штатов и составить их родословные. [ 120 ] Давенпорт использовал эту информацию, чтобы документировать различный возраст начала и диапазон симптомов ГБ; он утверждал, что большинство случаев ГБ в США можно отнести к небольшому числу людей. [ 120 ] Это исследование было дополнительно приукрашено в 1932 году П.Р. Весси , который популяризировал идею о том, что три брата, покинувшие Англию в 1630 году и направлявшиеся в Бостон, были прародителями ГБ в США. [ 121 ] Утверждение о том, что были установлены самые ранние прародители, и евгеническая предвзятость работ Манси, Давенпорта и Весси способствовали недопониманию и предрассудкам в отношении БГ. [ 113 ] Манси и Давенпорт также популяризировали идею о том, что в прошлом некоторые люди с HD могли считаться одержимыми духами или жертвами колдовства , и иногда их избегали или изгоняли общество. [ 122 ] [ 123 ] Эта идея не доказана. Исследователи нашли противоположные доказательства; например, сообщество семьи, которую изучал Джордж Хантингтон, открыто принимало тех, у кого были симптомы ГБ. [ 113 ] [ 122 ]
Поиск причины этого состояния значительно активизировался в 1968 году, когда Фонд наследственных заболеваний был создан Милтоном Векслером , психоаналитиком из Лос-Анджелеса , Калифорния , у чьей жены Леоноры Сабин ранее в том же году была диагностирована болезнь Хантингтона, (HDF). . [ 124 ] Трое братьев жены Векслера также страдали этим заболеванием.
Фонд участвовал в привлечении более 100 ученых для участия в совместном проекте США и Венесуэлы по борьбе с болезнью Хантингтона, который в течение 10 лет, начиная с 1979 года, работал над определением генетической причины. [ 125 ] Это было достигнуто в 1983 году, когда был приблизительно обнаружен причинный ген, [ 104 ] а в 1993 году этот ген был точно расположен на хромосоме 4 (4p16.3). [ 126 ] Исследование было сосредоточено на населении двух изолированных венесуэльских деревень, Барранкитас и Лагунетас, где наблюдалась необычно высокая распространенность ГБ, и охватило более 18 000 человек, в основном из одной большой семьи, и привело к тому, что ГБ стал первым аутосомного заболевания. локусом обнаружено с помощью анализа генетического сцепления . [ 126 ] [ 127 ] Среди других инноваций в рамках проекта были разработаны методы маркировки ДНК , которые стали важным шагом на пути к реализации проекта «Геном человека» . [ 125 ]
В то же время были сделаны ключевые открытия, касающиеся механизмов заболевания, в том числе выводы исследовательской группы Аниты Хардинг о влиянии длины гена. [ 128 ]
Моделирование заболевания на различных типах животных, таких как трансгенная мышь, выведенная в 1996 году, позволило провести более масштабные эксперименты. Поскольку у этих животных более быстрый метаболизм и гораздо более короткая продолжительность жизни, чем у людей, результаты экспериментов получаются раньше, что ускоряет исследования. Открытие в 1997 году того, что фрагменты mHtt неправильно сворачиваются, привело к открытию ядерных включений, которые они вызывают. Эти достижения привели к все более обширным исследованиям белков, связанных с этим заболеванием, потенциальных медикаментозных методов лечения, методов ухода и самого гена. [ 111 ] [ 129 ]
Сети ухода и поддержки, которые сложились в Венесуэле и Колумбии во время исследовательских проектов там в 1970-х и 2000-х годах, в конечном итоге были разрушены различными силами, такими как продолжающийся кризис в Венесуэле и смерть ведущего исследователя в Колумбии (Хорхе Даса Баррига). ). [ 130 ] Врачи работают над возрождением этих сетей, потому что люди, которые внесли свой вклад в науку о болезни Хантингтона, участвуя в этих исследованиях, заслуживают надлежащего последующего ухода; общества в других частях мира, которые извлекают выгоду из достигнутых таким образом научных достижений, обязаны по крайней мере этим тем, кто участвовал в исследованиях. [ 130 ]
Раньше это состояние называлось хореей Хантингтона, но этот термин был заменен болезнью Хантингтона, поскольку не у всех пациентов развивается хорея, а также из-за важности когнитивных и поведенческих проблем. [ 131 ]
Общество и культура
[ редактировать ]Этика
[ редактировать ]Генетическое тестирование болезни Хантингтона подняло несколько этических проблем. Проблемы генетического тестирования включают определение того, насколько зрелым должен быть человек, прежде чем его можно будет считать подходящим для тестирования, обеспечение конфиденциальности результатов и следует ли разрешить компаниям использовать результаты тестов для принятия решений о приеме на работу, страховании жизни или других финансовых вопросах. Возникли разногласия, когда в 1910 году Чарльз Дэвенпорт предложил использовать принудительную стерилизацию и иммиграционный контроль для людей с определенными заболеваниями, включая БГ, в рамках евгенического движения. [ 132 ] Экстракорпоральное оплодотворение имеет некоторые проблемы, связанные с использованием эмбрионов. Некоторые исследования БГ имеют этические проблемы из-за использования испытаний на животных и эмбриональных стволовых клеток . [ 133 ] [ 134 ]
Разработка точного диагностического теста на болезнь Хантингтона вызвала социальные, юридические и этические проблемы по поводу доступа к результатам человека и их использования. [ 135 ] [ 136 ] Многие руководства и процедуры тестирования предусматривают строгие процедуры раскрытия информации и конфиденциальности, позволяющие людям решать, когда и как получать результаты, а также кому они будут доступны. [ 26 ] Страховые компании и предприятия сталкиваются с вопросом, следует ли использовать результаты генетических тестов при оценке человека, например, при страховании жизни или трудоустройстве. Страховые компании Соединенного Королевства договорились с Министерством здравоохранения и социального обеспечения , что до 2017 года клиентам не нужно будет раскрывать им прогностические генетические тесты, но это соглашение прямо исключает одобренный правительством тест для болезни Хантингтона при оформлении полисов на сумму более 500 000 фунтов стерлингов. . [ 137 ] [ 138 ] Как и в случае с другими неизлечимыми генетическими заболеваниями с более поздним началом, проведение предсимптомного тестирования у ребенка или подростка является этически сомнительным, поскольку для этого человека не будет никакой медицинской пользы. Существует консенсус в отношении тестирования только лиц, которые считаются когнитивно зрелыми, хотя существует контраргумент, согласно которому родители имеют право принимать решение от имени своего ребенка. Из-за отсутствия эффективного лечения тестирование лица, не достигшего совершеннолетия , которое не признано дееспособным, в большинстве случаев считается неэтичным. [ 49 ] [ 139 ] [ 140 ]
Существуют этические проблемы, связанные с пренатальным генетическим тестированием или преимплантационной генетической диагностикой, чтобы гарантировать, что ребенок не родится с данным заболеванием. [ 141 ] Например, пренатальное тестирование поднимает вопрос о селективном аборте, который некоторые считают неприемлемым. [ 141 ] Поскольку это доминантное заболевание, возникают трудности в ситуациях, когда родитель не хочет знать свой диагноз. Это потребует сохранения частей процесса в секрете от родителя. [ 141 ]
Поддерживающие организации
[ редактировать ]
В 1968 году, после того как Милтон Векслер столкнулся с HD в семье своей жены, он был вдохновлен созданием Фонда наследственных заболеваний (HDF) с целью лечения генетических заболеваний путем координации и поддержки исследований. [ 17 ] Фонд и дочь Векслера, Нэнси Векслер , были ключевыми участниками исследовательской группы в Венесуэле, которая обнаружила ген HD. [ 17 ]
Примерно в то же время, когда сформировалась HDF, Марджори Гатри помогла основать комитет по борьбе с болезнью Хантингтона (ныне Американское общество по борьбе с болезнью Хантингтона ) после того, как ее муж, фолк-певец и автор песен Вуди Гатри умер от осложнений HD. [ 18 ]
С тех пор во многих странах мира сформировались организации поддержки и исследования, которые помогли повысить осведомленность общественности о БГ. Некоторые из них сотрудничают в зонтичных организациях, таких как Международная ассоциация Хантингтона и Европейская сеть HD. [ 142 ] Многие организации поддержки проводят ежегодные мероприятия по повышению осведомленности о БГ, некоторые из которых были одобрены соответствующими правительствами. Например, Сенат США объявил 6 июня «Национальным днем распространения информации о болезни Хантингтона» . [ 143 ] Существует множество организаций, которые поддерживают и информируют людей, страдающих ГБ, включая Ассоциацию болезни Хантингтона в Великобритании. Крупнейшим спонсором исследований является Фонд инициативы по лечению болезни Хантингтона (CHDI). [ 144 ]
Направления исследований
[ редактировать ]Исследования механизма ГБ сосредоточены на выявлении функционирования Htt, того, как mHtt отличается или мешает ему, а также патологии головного мозга, вызываемой этим заболеванием. [ 145 ] Исследования проводятся с использованием in vitro методов , генетически модифицированных животных (также называемых моделями трансгенных животных ) и людей-добровольцев. Модели на животных имеют решающее значение для понимания фундаментальных механизмов, вызывающих заболевание, а также для поддержки ранних стадий разработки лекарств . [ 129 ] Идентификация причинного гена позволила создать множество генетически модифицированных организмов , включая нематод (круглых червей), -дрозофил плодовых мух и генетически модифицированных млекопитающих, включая мышей, крыс, овец, свиней и обезьян, которые экспрессируют мутантный хантингтин и развивают прогрессирующую нейродегенерацию и HD-ген. как симптомы. [ 129 ]
В настоящее время проводятся исследования с использованием множества подходов, направленных либо на предотвращение болезни Хантингтона, либо на замедление ее прогрессирования. [ 145 ] Стратегии, модифицирующие заболевание, можно разделить на три категории: снижение уровня мутантного белка хантингтина (включая сплайсинг генов и подавление генов ); подходы, направленные на улучшение выживаемости нейронов за счет снижения вреда, наносимого белком конкретным клеточным путям и механизмам (включая гомеостаз белка и ингибирование деацетилазы гистонов ); и стратегии замены потерянных нейронов. Кроме того, разрабатываются новые методы лечения для улучшения функционирования мозга; они направлены на разработку симптоматических, а не модифицирующих заболевание методов лечения и включают ингибиторы фосфодиэстеразы . [ 146 ] [ 147 ]
Фонд CHDI финансирует множество исследовательских инициатив, публикуя множество публикаций. [ 148 ] Фонд CHDI является крупнейшим спонсором исследований болезни Хантингтона в мире и стремится найти и разработать лекарства, которые замедлят прогрессирование ГБ. [ 144 ] [ 149 ] CHDI ранее был известен как High Q Foundation. В 2006 году компания потратила 50 миллионов долларов на исследования болезни Хантингтона. [ 144 ] CHDI сотрудничает со многими академическими и коммерческими лабораториями по всему миру и занимается надзором и управлением исследовательскими проектами, а также финансированием. [ 150 ]
Сокращение производства хантингтина
[ редактировать ]Замалчивание генов направлено на снижение продукции мутантного белка, поскольку БХ вызывается единственным доминантным геном, кодирующим токсичный белок. Эксперименты по подавлению генов на моделях мышей показали, что когда экспрессия mHtt снижается, симптомы улучшаются. [ 151 ] Безопасность РНК-интерференции и методов подавления генов с помощью аллель-специфических олигонуклеотидов (ASO) была продемонстрирована на мышах и мозге более крупных приматов-макак. [ 152 ] [ 153 ] Аллель-специфическое подавление пытается заставить замолчать мутантный htt, оставляя Htt дикого типа нетронутым. Один из способов добиться этого — идентифицировать полиморфизмы, присутствующие только в одной аллели, и производить препараты, подавляющие гены, которые нацелены на полиморфизмы только в мутантном аллеле. [ 154 ] Первое испытание по подавлению генов с участием людей с БГ началось в 2015 году с целью проверки безопасности IONIS-HTTRx, произведенного Ionis Pharmaceuticals и возглавляемого Институтом неврологии UCL . [ 155 ] [ 156 ] Мутантный хантингтин был впервые обнаружен и количественно определен в спинномозговой жидкости носителей мутации болезни Хантингтона в 2015 году с использованием нового иммуноанализа «подсчет одиночных молекул» . [ 157 ] предоставляя прямой способ оценить, достигают ли методы снижения хантингтина желаемого эффекта. [ 158 ] [ 159 ] Испытание третьей фазы этого соединения, переименованного в томинерсен и спонсируемого Roche Pharmaceuticals , началось в 2019 году, но было остановлено в 2021 году после того, как совет по мониторингу безопасности пришел к выводу, что баланс риска и пользы был неблагоприятным. [ 160 ] Испытания генной терапии, снижающие хантингтин, проводимые Uniqure, начались в 2019 году, и было объявлено о нескольких испытаниях пероральных соединений-модуляторов сплайсинга, снижающих хантингтин. [ 161 ] сплайсинга генов Рассматриваются методы , чтобы попытаться восстановить геном с ошибочным геном, вызывающим БГ, с использованием таких инструментов, как CRISPR/Cas9 . [ 147 ]
Увеличение клиренса гентингтина
[ редактировать ]Другая стратегия снижения уровня мутантного хантингтина заключается в увеличении скорости, с которой клетки способны его выводить. [ 162 ] Поскольку mHtt (и многие другие белковые агрегаты ) разрушаются в результате аутофагии , увеличение скорости аутофагии потенциально может снизить уровни mHtt и тем самым облегчить течение заболевания. [ 163 ] Фармакологические и генетические индукторы аутофагии были протестированы на различных моделях болезни Хантингтона; Было показано, что многие из них снижают уровни mHtt и снижают токсичность. [ 162 ]
Улучшение выживаемости клеток
[ редактировать ]Среди подходов, направленных на улучшение выживаемости клеток в присутствии мутантного хантингтина, — коррекция регуляции транскрипции с помощью ингибиторов гистондеацетилазы , модуляция агрегации хантингтина, улучшение метаболизма и функции митохондрий и восстановление функции синапсов . [ 151 ]
Замена нейронов
[ редактировать ]Терапия стволовыми клетками используется для замены поврежденных нейронов путем трансплантации стволовых клеток в пораженные участки мозга. Эксперименты на животных моделях (только крысы и мыши) дали положительные результаты. [ 164 ]
Каким бы ни был их будущий терапевтический потенциал, стволовые клетки уже являются ценным инструментом для изучения болезни Хантингтона в лаборатории. [ 165 ]
Ферроптоз
[ редактировать ]Ферроптоз — это форма регулируемой гибели клеток, характеризующаяся зависимым от железа накоплением гидроперекисей липидов до летальных уровней. ALOX5 -опосредованный ферроптоз действует как путь гибели клеток при окислительном стрессе при болезни Хантингтона. [ 166 ] Ингибиторы ферроптоза защищают модели дегенеративных заболеваний головного мозга, в том числе Болезни Паркинсона, Хантингтона и Альцгеймера. [ 166 ]
Клинические испытания
[ редактировать ]В 2020 году было проведено 197 клинических исследований, связанных с различными методами лечения и биомаркерами болезни Хантингтона, которые были указаны как находящиеся в стадии реализации, набор или недавно завершенные. [ 167 ] соединения Испытанные , которые не смогли предотвратить или замедлить прогрессирование болезни Хантингтона, включают ремасемид , коэнзим Q10 , рилузол , креатин , миноциклин , этил-ЭПК , фенилбутират и димебон . [ 168 ]
См. также
[ редактировать ]Ссылки
[ редактировать ]- ^ Jump up to: а б с д и ж г час я дж к л Даялу П., Альбин Р.Л. (февраль 2015 г.). «Болезнь Хантингтона: патогенез и лечение». Неврологические клиники . 33 (1): 101–114. дои : 10.1016/j.ncl.2014.09.003 . ПМИД 25432725 .
- ^ Jump up to: а б с д и ж г Кэрон Н.С., Райт Дж.Э., Хайден М.Р. (2020). Адам М.П., Ардингер Х.Х., Пагон Р.А., Уоллес С.Е., Бин Л.Дж., Стивенс К., Амемия А. (ред.). «Болезнь Хантингтона». Джин Обзоры . ПМИД 20301482 .
- ^ Jump up to: а б с д и ж г час я дж к л м Фрэнк С. (январь 2014 г.). «Лечение болезни Гентингтона» . Нейротерапия . 11 (1): 153–160. дои : 10.1007/s13311-013-0244-z . ПМЦ 3899480 . ПМИД 24366610 .
- ^ Jump up to: а б с д и ж г час «Информационная страница о болезни Хантингтона» . Национальный институт неврологических расстройств и инсульта . Архивировано из оригинала 13 декабря 2020 года . Проверено 14 декабря 2020 г.
- ^ Jump up to: а б Дурр А., Гарджуло М., Фейнгольд Дж. (ноябрь 2012 г.). «Пресимптоматическая фаза болезни Гентингтона». Ревю Неврологии . 168 (11): 806–808. дои : 10.1016/j.neurol.2012.07.003 . ПМИД 22902173 .
- ^ Ферри ФФ (2010). Дифференциальный диагноз Ферри: практическое руководство по дифференциальной диагностике симптомов, признаков и клинических нарушений (2-е изд.). Филадельфия, Пенсильвания: Эльзевир/Мосби. п. Глава H. ISBN 978-0-323-07699-9 .
- ^ «Болезнь Хантингтона – Лечение и поддержка» . Национальная служба здравоохранения Великобритании . 23 октября 2017 года. Архивировано из оригинала 6 мая 2023 года . Проверено 6 мая 2023 г.
- ^ Jump up to: а б Illarioshkin SN, Klyushnikov SA, Vigont VA, Seliverstov YA, Kaznacheyeva EV (September 2018). "Molecular Pathogenesis in Huntington's Disease" . Biochemistry. Biokhimiia . 83 (9): 1030–1039. doi : 10.1134/S0006297918090043 . PMID 30472941 . S2CID 26471825 . Archived from the original on 13 November 2020 . Retrieved 8 November 2020 – via protein.bio.msu.ru.
- ^ Jump up to: а б с Судхакар В., Ричардсон Р.М. (январь 2019 г.). «Генная терапия нейродегенеративных заболеваний» . Нейротерапия . 16 (1): 166–175. дои : 10.1007/s13311-018-00694-0 . ПМК 6361055 . ПМИД 30542906 .
- ^ Jump up to: а б с Кумар, Аббас А., Астер Дж. (2018). Основная патология Роббинса (Десятое изд.). Филадельфия, Пенсильвания: Эльзевир. п. 879. ИСБН 978-0-323-35317-5 .
- ^ Первс Д. (2012). Нейронаука (5-е изд.). Сандерленд, Массачусетс: Sinauer Associates. п. 415. ИСБН 978-0-87893-695-3 .
- ^ Jump up to: а б с Сауду Ф., Умберт С. (март 2016 г.). «Биология Хантингтина» . Нейрон . 89 (5): 910–926. дои : 10.1016/j.neuron.2016.02.003 . ПМИД 26938440 . S2CID 8272667 .
- ^ Jump up to: а б с д и Бейтс Г.П., Дорси Р., Гуселла Дж.Ф., Хайден М.Р., Кей С., Ливитт Б.Р. и др. (апрель 2015 г.). «Болезнь Хантингтона». Обзоры природы. Праймеры по болезням . 1 : 15005. дои : 10.1038/nrdp.2015.5 . ПМИД 27188817 . S2CID 25759303 .
- ^ «Аспирационная пневмония: что это такое, причины, диагностика, лечение» . Кливлендская клиника . Архивировано из оригинала 12 июня 2023 года . Проверено 12 июня 2023 г.
- ^ Jump up to: а б Вейл ТК, Кардосо Ф (2015). «Хорея: Путешествие в историю» . Тремор и другие гиперкинетические движения . 5 . дои : 10.7916/D8WM1C98 . ПМЦ 4454991 . ПМИД 26056609 .
- ^ Jump up to: а б «О болезни Хантингтона» . Genome.gov . Архивировано из оригинала 9 января 2021 года . Проверено 13 января 2021 г.
- ^ Jump up to: а б с д «История ХДФ» . Фонд наследственных болезней. Архивировано из оригинала 19 ноября 2015 года . Проверено 18 ноября 2015 г.
- ^ Jump up to: а б «История и генетика болезни Хантингтона | Американское общество болезни Хантингтона» . Март 2019. Архивировано из оригинала 1 декабря 2020 года . Проверено 14 декабря 2020 г.
- ^ Jump up to: а б Йенсен Р.Н., Болвиг Т., Соренсен С.А. (март 2018 г.). «[Психические симптомы у пациентов с болезнью Гентингтона]». Ugeskrift для Laeger (на датском языке). 180 (13). ПМИД 29587954 .
- ^ Jump up to: а б «Болезнь Хантингтона – Симптомы и причины» . Клиника Мэйо . Архивировано из оригинала 5 марта 2018 года . Проверено 13 декабря 2022 г.
- ^ «Болезнь Хантингтона» . www.nhsinform.scot. Архивировано из оригинала 12 июля 2020 года . Проверено 12 июля 2020 г.
- ^ Jump up to: а б с Кэрон Н.С., Райт Дж.Э., Хайден М.Р. (июнь 2020 г.). «Болезнь Хантингтона» . Книжная полка Genereviews . Университет Вашингтона. ПМИД 20301482 . Архивировано из оригинала 10 февраля 2009 года . Проверено 22 ноября 2020 г.
- ^ Диагностическое и статистическое руководство по психическим расстройствам: ДСМ-5 (5-е изд.). Арлингтон, Вирджиния: Американская психиатрическая ассоциация. 2013. с. 639. ИСБН 978-0-89042-554-1 .
- ^ Кремер Б. (2002). «Клиническая неврология болезни Гентингтона». В Бейтс Г., Харпер П., Джонс Л. (ред.). Болезнь Хантингтона – Третье издание . Оксфорд: Издательство Оксфордского университета. стр. 28–53. ISBN 978-0-19-851060-4 .
- ^ Wagle AC, Wagle SA, Маркова И.С., Берриос Г.Е. (2000). «Психиатрическая заболеваемость при болезни Гентингтона». Неврология, психиатрия и исследования мозга (8): 5–16.
- ^ Jump up to: а б с д и ж г час я дж к л м н тот п д р с т в v В х и С аа аб и объявление но из в ах есть также и аль являюсь а к ап ак с как в В из хорошо топор является тот нет бб до нашей эры др. быть парень бг чб Уокер Ф.О. (январь 2007 г.). «Болезнь Хантингтона». Ланцет . 369 (9557): 218–228. дои : 10.1016/S0140-6736(07)60111-1 . ПМИД 17240289 . S2CID 46151626 .
- ^ Кремер Б. (2002). «Клиническая неврология болезни Гентингтона». В Бейтс Г., Харпер П., Джонс Л. (ред.). Болезнь Хантингтона – Третье издание . Оксфорд: Издательство Оксфордского университета. стр. 28–53. ISBN 978-0-19-851060-4 .
- ^ Jump up to: а б с д Монтойя А., Прайс Б.Х., Менеар М., Лепаж М. (январь 2006 г.). «Визуализация мозга и когнитивные дисфункции при болезни Хантингтона» (PDF) . Журнал психиатрии и неврологии . 31 (1): 21–29. ПМК 1325063 . ПМИД 16496032 . Архивировано из оригинала (PDF) 23 марта 2016 года . Проверено 17 сентября 2008 г.
- ^ Jump up to: а б Дики А.С., Ла Спада, Арканзас (апрель 2018 г.). «Развитие терапии болезни Хантингтона: от текущих стратегий к новым возможностям» . Американский журнал медицинской генетики. Часть А. 176 (4): 842–861. дои : 10.1002/ajmg.a.38494 . ПМЦ 5975251 . ПМИД 29218782 .
- ^ Азиз Н.А., ван дер Марк М.А., Пейл Х., Олде Риккерт М.Г., Блум Б.Р., Роос Р.А. (декабрь 2008 г.). «Потеря веса при нейродегенеративных заболеваниях». Журнал неврологии . 255 (12): 1872–1880. дои : 10.1007/s00415-009-0062-8 . ПМИД 19165531 . S2CID 26109381 .
- ^ «Брошюра Канадского общества Хантингтона» (PDF) . Справочник для лиц, осуществляющих уход за болезнью Гентингтона на поздней стадии . Общество HD Канады. 11 апреля 2007 г. Архивировано из оригинала (PDF) 25 июня 2008 г. . Проверено 10 августа 2008 г.
- ^ Jump up to: а б с ван Дуйн Э., Кингма Э.М., ван дер Маст Р.К. (2007). «Психопатология у подтвержденных носителей гена болезни Хантингтона». Журнал нейропсихиатрии и клинических нейронаук . 19 (4): 441–448. дои : 10.1176/appi.neuropsych.19.4.441 . ПМИД 18070848 .
- ^ Мюррей Э.Д., Баттнер Н., Прайс Б.Х. (2012). «Депрессия и психоз в неврологической практике». В Брэдли В.Г., Дарофф Р.Б., Феничел Г.М., Янкович Дж. (ред.). Неврология Брэдли в клинической практике (6-е изд.). Филадельфия, Пенсильвания: Эльзевир/Сондерс. п. 108. ИСБН 978-1-4377-0434-1 .
- ^ ван дер Бург Дж. М., Бьоркквист М., Брундин П. (август 2009 г.). «За пределами мозга: широко распространенная патология при болезни Хантингтона». «Ланцет». Неврология . 8 (8): 765–774. дои : 10.1016/S1474-4422(09)70178-4 . ПМИД 19608102 . S2CID 14419437 .
- ^ Кацуно М., Банно Х., Сузуки К., Такеучи Ю., Кавасима М., Танака Ф. и др. (май 2008 г.). «Молекулярная генетика и биомаркеры полиглутаминовых заболеваний». Современная молекулярная медицина . 8 (3): 221–234. дои : 10.2174/156652408784221298 . ПМИД 18473821 .
- ^ Сквитьери Ф, Фрати Л, Чармиелло А, Ластория С, Куоррел О (февраль 2006 г.). «Ювенильная болезнь Хантингтона: отличается ли дозо-эффект патогенный механизм от классической болезни взрослых?». Механизмы старения и развития . 127 (2): 208–212. дои : 10.1016/j.mad.2005.09.012 . ПМИД 16274727 . S2CID 20523093 .
- ^ Нэнс М.А., Майерс Р.Х. (2001). «Ювенильное начало болезни Хантингтона - клинические и исследовательские перспективы». Обзоры исследований умственной отсталости и нарушений развития . 7 (3): 153–157. дои : 10.1002/mrdd.1022 . ПМИД 11553930 .
- ^ Пассаж Е (2001). Цветной атлас генетики (2-е изд.). Темы. п. 142 . ISBN 978-0-86577-958-7 .
- ^ «Секс связан» . Genome.gov . Архивировано из оригинала 14 апреля 2022 года . Проверено 13 декабря 2022 г.
- ^ Ридли Р.М., Фрит К.Д., Кроу Т.Дж., Коннели П.М. (сентябрь 1988 г.). «Предчувствие при болезни Гентингтона наследуется по мужской линии, но может возникать и по женской линии» . Журнал медицинской генетики . 25 (9): 589–595. дои : 10.1136/jmg.25.9.589 . ПМЦ 1051535 . ПМИД 2972838 .
- ^ Семака А., Крейтон С., Уорби С., Хайден М.Р. (октябрь 2006 г.). «Прогностическое тестирование на болезнь Хантингтона: интерпретация и значение промежуточных аллелей». Клиническая генетика . 70 (4): 283–294. дои : 10.1111/j.1399-0004.2006.00668.x . ПМИД 16965319 . S2CID 26007984 .
- ^ Векслер Н.С., Янг А.Б., Танци Р.Э., Трэверс Х., Староста-Рубинштейн С., Пенни Дж.Б. и др. (1987). «Гомозиготы по болезни Гентингтона». Природа . 326 (6109): 194–197. Бибкод : 1987Natur.326..194W . дои : 10.1038/326194a0 . hdl : 2027.42/62543 . ПМИД 2881213 . S2CID 4312171 .
- ^ Сквитьери Ф., Геллера С., Каннелла М., Мариотти С., Цислаги Г., Рубинштейн Д.С. и др. (апрель 2003 г.). «Гомозиготность по мутации CAG при болезни Хантингтона связана с более тяжелым клиническим течением» . Мозг . 126 (Часть 4): 946–955. дои : 10.1093/brain/awg077 . ПМИД 12615650 .
- ^ Гёлер Х., Лаловски М., Стельцль У., Вельтер С., Стродике М., Ворм У. и др. (сентябрь 2004 г.). «Сеть взаимодействия белков связывает GIT1, усилитель агрегации хантингтина, с болезнью Хантингтона» . Молекулярная клетка . 15 (6): 853–865. doi : 10.1016/j.molcel.2004.09.016 . ПМИД 15383276 .
- ^ Глайч К.Е., Садри-Вакили Г (2015). «Эпигенетические механизмы, участвующие в патогенезе болезни Хантингтона» . Журнал болезни Хантингтона . 4 (1): 1–15. дои : 10.3233/JHD-159001 . ПМИД 25813218 .
- ^ Jump up to: а б Лю З, Чжоу Т, Зиглер А.С., Димитрион П., Цзо Л. (2017). «Окислительный стресс при нейродегенеративных заболеваниях: от молекулярных механизмов к клиническому применению» . Окислительная медицина и клеточное долголетие . 2017 : 2525967. дои : 10.1155/2017/2525967 . ПМК 5529664 . ПМИД 28785371 .
- ^ Jump up to: а б с Каттанео Э., Зуккато С., Тартари М. (декабрь 2005 г.). «Нормальная функция хантингтина: альтернативный подход к болезни Гентингтона». Обзоры природы. Нейронаука . 6 (12): 919–930. дои : 10.1038/nrn1806 . ПМИД 16288298 . S2CID 10119487 .
- ^ Jump up to: а б с д и Рубинштейн, округ Колумбия, Кармайкл Дж (август 2003 г.). «Болезнь Хантингтона: молекулярная основа нейродегенерации». Обзоры экспертов в области молекулярной медицины . 5 (20): 1–21. дои : 10.1017/S1462399403006549 . ПМИД 14585171 . S2CID 28435830 .
- ^ Jump up to: а б Блох М., Хайден М.Р. (январь 1990 г.). «Мнение: прогностическое тестирование на болезнь Хантингтона в детстве: проблемы и последствия» . Американский журнал генетики человека . 46 (1): 1–4. ПМК 1683548 . ПМИД 2136787 .
- ^ Матлахов И., ван дер Вель ПК (декабрь 2019 г.). «Конформационные исследования патогенных расширенных полиглутаминовых белковых отложений при болезни Хантингтона» . Экспериментальная биология и медицина . 244 (17): 1584–1595. дои : 10.1177/1535370219856620 . ПМК 6920524 . ПМИД 31203656 . S2CID 189944779 .
- ^ Jump up to: а б с Садри-Вакили Г., Ча Дж. Х. (июнь 2006 г.). «Механизмы заболевания: модификации гистонов при болезни Хантингтона». Природная клиническая практика. Неврология . 2 (6): 330–338. дои : 10.1038/ncpneuro0199 . ПМИД 16932577 . S2CID 12474262 .
- ^ Кумар А., Ратан Р.Р. (октябрь 2016 г.). «Окислительный стресс и болезнь Хантингтона: хорошее, плохое и злое» . Журнал болезни Хантингтона . 5 (3): 217–237. дои : 10.3233/JHD-160205 . ПМК 5310831 . ПМИД 27662334 .
- ^ ПК Нопулос (март 2016 г.). «Болезнь Хантингтона: моногенное дегенеративное заболевание полосатого тела» . Диалоги в клинической неврологии . 18 (1): 91–98. doi : 10.31887/DCNS.2016.18.1/pnopoulos . ПМЦ 4826775 . ПМИД 27069383 .
- ^ МакКолган П., Тебризи С.Дж. (январь 2018 г.). «Болезнь Хантингтона: клинический обзор» . Европейский журнал неврологии . 25 (1): 24–34. дои : 10.1111/ene.13413 . ПМИД 28817209 .
- ^ Первс Д., Августин Г.А., Фитцпатрик Д., Холл В., ЛаМантия А.С., Макнамара Д.О. и др. (2001). «Модуляция движения базальных ганглиев - цепи в системе базальных ганглиев» . В Первес Д. (ред.). Нейронаука (2-е изд.). Сандерленд, Массачусетс: Sinauer Associates. ISBN 978-0-87893-742-4 . Архивировано из оригинала 18 февраля 2009 года . Проверено 1 апреля 2009 г.
- ^ Лобсигер CS, Кливленд DW (ноябрь 2007 г.). «Глиальные клетки как внутренние компоненты неклеточных автономных нейродегенеративных заболеваний» . Природная неврология . 10 (11): 1355–1360. дои : 10.1038/nn1988 . ПМК 3110080 . ПМИД 17965655 .
- ^ Jump up to: а б Кроссман А.Р. (май 2000 г.). «Функциональная анатомия двигательных нарушений» . Журнал анатомии . 196 (Часть 4): 519–525. дои : 10.1046/j.1469-7580.2000.19640519.x . ПМК 1468094 . ПМИД 10923984 .
- ^ Даффи Дж (2013). Двигательные нарушения речи: субстраты, дифференциальный диагноз и лечение (3-е изд.). Сент-Луис, Миссури: Эльзевир. стр. 196–7.
- ^ Стеффан Дж.С., Бодай Л., Паллос Дж., Поулман М., МакКэмпбелл А., Апостол Б.Л. и др. (октябрь 2001 г.). «Ингибиторы гистондеацетилазы останавливают полиглутамин-зависимую нейродегенерацию у дрозофилы» . Природа . 413 (6857): 739–743. Бибкод : 2001Natur.413..739S . дои : 10.1038/35099568 . ПМИД 11607033 . S2CID 4419980 . Архивировано из оригинала 1 августа 2020 года . Проверено 28 июня 2019 г.
- ^ Петрушка Дж., Хартенстайн М.Дж., Гудман М.Ф. (февраль 1998 г.). «Анализ проскальзывания цепи в расширениях ДНК-полимеразы триплетных повторов CAG/CTG, связанных с нейродегенеративными заболеваниями» . Журнал биологической химии . 273 (9): 5204–5210. дои : 10.1074/jbc.273.9.5204 . ПМИД 9478975 .
- ^ Гайяр Ф (1 мая 2007 г.). «Болезнь Хантингтона» . Радиологическая картина дня . www.radpod.org. Архивировано из оригинала 22 октября 2007 года . Проверено 24 июля 2009 г.
- ^ Рао АК, Муратори Л, Луис ЭД, Московиц КБ, Мардер КС (апрель 2009 г.). «Клиническое измерение нарушений подвижности и равновесия при болезни Гентингтона: достоверность и оперативность». Походка и осанка . 29 (3): 433–436. дои : 10.1016/j.gaitpost.2008.11.002 . ПМИД 19111470 .
- ^ «Единая шкала оценки болезни Хантингтона (UHDRS)» . UHDRS и база данных . ГСГ. 1 февраля 2009 года. Архивировано из оригинала 11 августа 2015 года . Проверено 14 апреля 2009 г.
- ^ Майерс Р.Х. (апрель 2004 г.). «Генетика болезни Хантингтона» . НейроРкс . 1 (2): 255–262. дои : 10.1602/neurorx.1.2.255 . ПМК 534940 . ПМИД 15717026 .
- ^ Jump up to: а б с д и ж г де Ди-Смолдерс CE, де Верт GM, Либерс И, Тиббен А, Эверс-Кибумс Г (май 2013 г.). «Репродуктивные возможности будущих родителей в семьях с болезнью Гентингтона: клинические, психологические и этические размышления» . Обновление репродукции человека . 19 (3): 304–315. дои : 10.1093/humupd/dms058 . ПМИД 23377865 . де Ди-Смолдерс CE, де Верт GM, Либерс И, Тиббен А, Эверс-Кибумс Г (2013). «Репродуктивные возможности будущих родителей в семьях с болезнью Гентингтона: клинические, психологические и этические размышления» . Обновление репродукции человека . 19 (3): 304–315. дои : 10.1093/humupd/dms058 . ПМИД 23377865 .
- ^ Байг С.С., Стронг М., Россер Э., Тавернер Н.В., Глю Р., Медзибродска З. и др. (октябрь 2016 г.). «22 года прогностического тестирования болезни Хантингтона: опыт Британского консорциума по прогнозированию Хантингтона» . Европейский журнал генетики человека . 24 (10): 1396–1402. дои : 10.1038/ejhg.2016.36 . ПМК 5027682 . ПМИД 27165004 .
- ^ Форрест Кинан К., Симпсон С.А., Медзибродска З., Александр Д.А., Семпер Дж. (июнь 2013 г.). «Как партнеры узнают о риске болезни Хантингтона в парных отношениях?». Журнал генетического консультирования . 22 (3): 336–344. дои : 10.1007/s10897-012-9562-2 . ПМИД 23297124 . S2CID 15447709 .
- ^ Эрвин С., Уильямс Дж.К., Джул А.Р., Менгелинг М., Миллс Дж.А., Бомбард Ю. и др. (июль 2010 г.). «Восприятие, опыт и реакция на генетическую дискриминацию при болезни Хантингтона: международное исследование RESPOND-HD» . Американский журнал медицинской генетики. Часть B. Нейропсихиатрическая генетика . 153Б (5): 1081–1093. дои : 10.1002/ajmg.b.31079 . ПМЦ 3593716 . ПМИД 20468061 .
- ^ Берсон CM, Марки КР (сентябрь 2001 г.). «Проблемы генетического консультирования при прогностическом генетическом тестировании семейных неврологических заболеваний, возникающих во взрослом возрасте». Семинары по детской неврологии . 8 (3): 177–186. дои : 10.1053/spen.2001.26451 . ПМИД 11575847 .
- ^ Смит Дж. А., Мичи С., Стивенсон М., Куоррел О. (март 2002 г.). «Восприятие риска и процессы принятия решений у кандидатов на генетическое тестирование на болезнь Гентингтона: интерпретирующий феноменологический анализ». Журнал психологии здоровья . 7 (2): 131–144. дои : 10.1177/1359105302007002398 . ПМИД 22114233 . S2CID 40182214 .
- ^ Jump up to: а б Хайден М.Р. (март 2003 г.). «Прогностическое тестирование на болезнь Гентингтона: универсальная модель?». «Ланцет». Неврология . 2 (3): 141–142. дои : 10.1016/S1474-4422(03)00317-X . ПМИД 12849232 . S2CID 39581496 .
- ^ «Руководство по прогностическому тесту молекулярной генетики при болезни Хантингтона. Международная ассоциация Хантингтона (IHA) и Всемирная федерация неврологов (WFN) Исследовательская группа по хорее Гентингтона». Неврология . 44 (8): 1533–1536. Август 1994 г. doi : 10.1212/WNL.44.8.1533 . ПМИД 8058167 . S2CID 28018134 .
- ^ Лосекут М., ван Бельзен М.Дж., Сенека С., Бауэр П., Стенхаус С.А., Бартон Д.Э. (май 2013 г.). «Руководство по передовому опыту EMQN/CMGS по молекулярно-генетическому тестированию болезни Хантингтона» . Европейский журнал генетики человека . 21 (5): 480–486. дои : 10.1038/ejhg.2012.200 . ПМЦ 3641377 . ПМИД 22990145 .
- ^ Шульман Дж.Д., Блэк С.Х., Хэндисайд А., Нэнс В.Е. (февраль 1996 г.). «Преимплантационное генетическое тестирование на болезнь Хантингтона и некоторые другие доминантно наследуемые заболевания». Клиническая генетика . 49 (2): 57–58. дои : 10.1111/j.1399-0004.1996.tb04327.x . ПМИД 8740912 . S2CID 45703511 .
- ^ Стерн Х.Дж., Хартон Г.Л., Сиссон М.Е., Джонс С.Л., Фэллон Л.А., Торселл Л.П. и др. (июнь 2002 г.). «Неразглашаемая преимплантационная генетическая диагностика болезни Хантингтона». Пренатальная диагностика . 22 (6): 503–507. дои : 10.1002/pd.359 . ПМИД 12116316 . S2CID 33967835 .
- ^ «Прогностическое тестирование на болезнь Хантингтона» . 2011. Архивировано из оригинала 22 января 2013 года . Проверено 7 мая 2013 г.
- ^ Кулиев А, Верлинский Ю (апрель 2005 г.). «Преимплантационная диагностика: реалистичный вариант вспомогательной репродукции и генетической практики». Современное мнение в области акушерства и гинекологии . 17 (2): 179–183. дои : 10.1097/01.gco.0000162189.76349.c5 . ПМИД 15758612 . S2CID 9382420 .
- ^ «Руководство по генетическому тестированию болезни Гентингтона» . Фонд наследственных болезней. Архивировано из оригинала 26 июня 2015 года . Проверено 7 мая 2013 г.
- ^ Jump up to: а б Шнайдер С.А., Уокер Р.Х., Бхатия К.П. (сентябрь 2007 г.). «Синдромы, подобные болезни Хантингтона: что следует учитывать у пациентов с отрицательным тестом на ген болезни Гентингтона». Природная клиническая практика. Неврология . 3 (9): 517–525. дои : 10.1038/ncpneuro0606 . ПМИД 17805246 . S2CID 9052603 .
- ^ Фрэнк С., Янкович Дж. (март 2010 г.). «Достижения в фармакологическом лечении болезни Хантингтона» . Наркотики . 70 (5): 561–571. дои : 10.2165/11534430-000000000-00000 . ПМИД 20329804 . S2CID 42386743 . Архивировано из оригинала 8 октября 2011 года.
- ^ Jump up to: а б с Бонелли Р.М., Веннинг Г.К., Капфхаммер Х.П. (март 2004 г.). «Болезнь Хантингтона: современные методы лечения и будущие методы лечения». Международная клиническая психофармакология . 19 (2): 51–62. дои : 10.1097/00004850-200403000-00001 . ПМИД 15076012 . S2CID 1956458 .
- ^ Панагиотакис П.Х., ДиСарио Дж.А., Хильден К., Огара М., Фанг Дж.К. (2008). «Размещение трубки DPEJ предотвращает аспирационную пневмонию у пациентов из группы высокого риска». Питание в клинической практике . 23 (2): 172–175. дои : 10.1177/0884533608314537 . ПМИД 18390785 .
- ^ Jump up to: а б «Руководство по физиотерапии EHDN» (PDF) . Европейская рабочая группа по сетевой физиотерапии HD. Архивировано из оригинала (PDF) 4 марта 2016 года . Проверено 15 ноября 2015 г.
- ^ Куин Л., Буси М. (февраль 2012 г.). «Разработка рекомендаций по физиотерапии и классификаций лечения людей с болезнью Гентингтона» . Управление нейродегенеративными заболеваниями . 2 (1): 21–31. дои : 10.2217/nmt.11.86 . Архивировано из оригинала 9 августа 2020 года . Проверено 10 мая 2012 г.
- ^ Халил Х., Куинн Л., ван Дёрсен Р., Мартин Р., Россер А., Бусс М. (январь 2012 г.). «Приверженность использованию DVD с домашними упражнениями у людей с болезнью Хантингтона: точки зрения участников» . Физиотерапия . 92 (1): 69–82. дои : 10.2522/ptj.20100438 . ПМИД 21960468 .
- ^ Трэверс Э., Джонс К., Никол Дж. (март 2007 г.). «Оказание паллиативной помощи при болезни Хантингтона». Международный журнал паллиативного ухода . 13 (3): 125–130. дои : 10.12968/ijpn.2007.13.3.23274 . ПМИД 17505405 .
- ^ «FDA одобрило первый препарат для лечения хореи при болезни Гентингтона» . Управление по контролю за продуктами и лекарствами США. 15 августа 2008 года. Архивировано из оригинала 21 августа 2008 года . Проверено 10 августа 2008 г.
- ^ Коппен Э.М., Роос Р.А. (январь 2017 г.). «Современные фармакологические подходы к уменьшению хореи при болезни Хантингтона» . Наркотики . 77 (1): 29–46. дои : 10.1007/s40265-016-0670-4 . ПМК 5216093 . ПМИД 27988871 .
- ^ Морси С., Халил С.М., Дохейм М.Ф., Камель М.Г., Эль-Басиони Д.С., Ахмед Хасан Х.И. и др. (август 2019 г.). «Эффективность этил-ЭПК в лечении болезни Хантингтона: систематический обзор и метаанализ». Acta Neuropsychiatrica . 31 (4): 175–185. дои : 10.1017/neu.2019.11 . hdl : 10069/39427 . ПМИД 30890195 . S2CID 84183892 .
- ^ Центр исследований по оценке лекарств (17 июля 2019 г.). «В поисках поздней дискинезии: революционное определение и одобрение валбеназина» . FDA . Архивировано из оригинала 3 декабря 2020 года . Проверено 15 ноября 2020 г.
- ^ Лламас Э., Коюнку С., Ли Х.Дж., Верманн М., Гутьеррес-Гарсия Р., Данкен Н. и др. (ноябрь 2023 г.). «Экспрессия in planta человеческого полиQ-расширенного фрагмента хантингтина обнаруживает механизмы предотвращения агрегации белков, связанных с заболеванием» . Природное старение . 3 (11): 1345–1357. дои : 10.1038/s43587-023-00502-1 . ПМЦ 10645592 . ПМИД 37783816 .
- ^ А'Кампо Л.Е., Сплиетхофф-Камминга Н.Г., Роос Р.А. (2012). «Программа обучения пациентов по поводу болезни Гентингтона (ПКП-ГД)» . Дж. Хантингтонс Дис . 1 (1): 47–56. дои : 10.3233/JHD-2012-120002 . ПМИД 25063190 .
- ^ Харпер П. (2002). «Генетическое консультирование и предсимптомное тестирование». В Бейтс Г., Харпер П., Джонс Л. (ред.). Болезнь Хантингтона – Третье издание . Оксфорд: Издательство Оксфордского университета. стр. 198–242. ISBN 978-0-19-851060-4 .
- ^ Харпер П.С. (июнь 1999 г.). «Болезнь Хантингтона: клиническая, генетическая и молекулярная модель нарушений полиглутаминовых повторов» . Философские труды Лондонского королевского общества. Серия Б, Биологические науки . 354 (1386): 957–61. дои : 10.1098/rstb.1999.0446 . ПМЦ 1692597 . ПМИД 10434293 .
- ^ Эндрю С.Е., Голдберг Ю.П., Кремер Б., Телениус Х., Тейлманн Дж., Адам С. и др. (август 1993 г.). «Взаимосвязь между длиной повторов тринуклеотида (CAG) и клиническими особенностями болезни Хантингтона». Природная генетика . 4 (4): 398–403. дои : 10.1038/ng0893-398 . ПМИД 8401589 . S2CID 20645822 .
- ^ Jump up to: а б Уокер Ф.О. (январь 2007 г.). «Болезнь Хантингтона». Ланцет . 369 (9557): 218–28. дои : 10.1016/S0140-6736(07)60111-1 . ПМИД 17240289 . S2CID 46151626 .
- ^ Кроуфорд Д., Сноуден Дж. (2002). «Нейропсихологические и нервно-психические аспекты болезни Хантингтона». В Бейтс Г., Харпер П., Джонс Л. (ред.). Болезнь Хантингтона – Третье издание . Оксфорд: Издательство Оксфордского университета. стр. 62–87. ISBN 978-0-19-851060-4 .
- ^ Ди Майо Л., Сквитьери Ф., Наполитано Дж., Кампанелла Дж., Трофаттер Дж.А., Коннелли П.М. (апрель 1993 г.). «Суицидальный риск при болезни Хантингтона» . Журнал медицинской генетики . 30 (4): 293–5. дои : 10.1136/jmg.30.4.293 . ПМЦ 1016335 . ПМИД 8487273 .
- ^ Jump up to: а б с д и ж Харпер П. (2002). «Эпидемиология болезни Хантингтона». В Бейтс Г., Харпер П., Джонс Л. (ред.). Болезнь Хантингтона – Третье издание . Оксфорд: Издательство Оксфордского университета. стр. 159–189. ISBN 978-0-19-851060-4 .
- ^ Шарон И., Шэрон Р., Уилкенс Дж.П., Эрсан Т. (2010). «Деменция болезни Хантингтона» . медицина, WebMD . Медскейп. Архивировано из оригинала 5 марта 2010 года . Проверено 16 мая 2010 г.
- ^ Драйвер-Данкли Э., Кэвинесс Дж. Н. (2007). «Болезнь Хантингтона». В Шапира А.Х. (ред.). Неврология и клиническая неврология . Мосби Эльзевир. стр. 879–885. ISBN 978-0-323-03354-1 .
- ^ Эванс С.Дж., Дуглас И., Роулинз М.Д., Векслер Н.С., Тебризи С.Дж., Смит Л. (октябрь 2013 г.). «Распространенность болезни Хантингтона у взрослых в Великобритании на основе диагнозов, зафиксированных в записях общей практики» . Журнал неврологии, нейрохирургии и психиатрии . 84 (10): 1156–60. дои : 10.1136/jnnp-2012-304636 . ПМЦ 3786631 . ПМИД 23482661 .
- ^ Авила-Жироо Р. (1973). «Медицинские и социальные аспекты хореи Хантингтона в штате Сулия, Венесуэла». Достижения в неврологии . 1 : 261–6. ISSN 0091-3952 . НАИД 10021247802.
- ^ Jump up to: а б Гуселла Дж.Ф., Векслер Н.С., Коннели П.М., Нейлор С.Л., Андерсон М.А., Танзи Р.Э. и др. (1983). «Полиморфный ДНК-маркер, генетически связанный с болезнью Хантингтона». Природа . 306 (5940): 234–8. Бибкод : 1983Natur.306..234G . дои : 10.1038/306234a0 . ПМИД 6316146 . S2CID 4320711 .
- ^ Сквитьери Ф., Эндрю С.Э., Голдберг Ю.П., Кремер Б., Спенс Н., Зейслер Дж. и др. (декабрь 1994 г.). «Анализ гаплотипов ДНК болезни Хантингтона открывает ключ к разгадке происхождения и механизмов распространения CAG, а также причин географических различий распространенности». Молекулярная генетика человека . 3 (12): 2103–14. дои : 10.1093/hmg/3.12.2103 . ПМИД 7881406 .
- ^ Свейнссон О, Халлдорссон С, Олафссон Э (июль 2012 г.). «Необычайно низкая распространенность болезни Хантингтона в Исландии». Европейская неврология . 68 (1): 48–51. дои : 10.1159/000337680 . ПМИД 22722209 . S2CID 207551998 .
- ^ Сипиля Ю., Хиетала М., Сийтонен А., Пяйваринта М., Маямаа К. (январь 2015 г.). «Эпидемиология болезни Хантингтона в Финляндии». Паркинсонизм и связанные с ним расстройства . 21 (1): 46–9. дои : 10.1016/j.parkreldis.2014.10.025 . ПМИД 25466405 .
- ^ Альмквист Э.В., Элтерман Д.С., Маклеод П.М., Хайден М.Р. (сентябрь 2001 г.). «Высокий уровень заболеваемости и отсутствие семейного анамнеза у четверти пациентов, у которых впервые диагностирована болезнь Хантингтона в Британской Колумбии». Клиническая генетика . 60 (3): 198–205. дои : 10.1034/j.1399-0004.2001.600305.x . ПМИД 11595021 . S2CID 19786394 .
- ^ Jump up to: а б Хантингтон Дж. (1872 г.). «О Хорея» . Медицинский и хирургический репортер Филадельфии . 26 (15): 317–321. ISBN 978-90-6186-011-2 . Архивировано из оригинала 21 апреля 2022 года . Проверено 21 апреля 2022 г.
- ^ Данглисон Р. (1842 г.). Медицинская практика... Том. 2. Филадельфия, Пенсильвания, США: Леа и Бланшар. стр. 312–313. Архивировано из оригинала 20 апреля 2022 года . Проверено 20 апреля 2022 г.
- ^ Jump up to: а б с д и ж г час я дж Харпер П. (2002). «Болезнь Хантингтона: историческая справка». В Бейтс Г., Харпер П., Джонс Л. (ред.). Болезнь Хантингтона – Третье издание . Оксфорд: Издательство Оксфордского университета. стр. 3–24. ISBN 978-0-19-851060-4 .
- ^ Горман CR (1846 г.). О форме хореи, в просторечии называемой Magrums (докторская диссертация). Филадельфия: Медицинский колледж Джефферсона.
- «Каталог Медицинского колледжа Джефферсона в Филадельфии: сессия 1846–1847 годов» . п. 14. Архивировано из оригинала 26 июня 2022 года.
- Данглисон Р. (1848). Медицинская практика... Том. 2 (3-е изд.). Филадельфия, Пенсильвания, США: Леа и Бланшар. п. 218.
- Бойд В.А. (6 ноября 1913 г.). «Наследственная хорея с сообщением о случае» . Бостонский медицинский и хирургический журнал . 169 (19): 680–685. дои : 10.1056/NEJM191311061691904 . Архивировано из оригинала 2 августа 2023 года . Проверено 21 апреля 2022 г. См. стр. 680.
- ^ Jump up to: а б с д Векслер А, Векслер Н (2008). Женщина, вошедшая в море. Хантингтон и возникновение генетической болезни . Издательство Йельского университета. п. 288. ИСБН 978-0-300-10502-5 . Проверено 15 ноября 2015 г.
- ^ Лунд Дж. К. (1860). «Хорея Сти Вити в Сетерсдалене. Отрывок из медицинского отчета окружного доктора Дж. К. Лунда за 1860 год». Отчет о состоянии здоровья : 137–138.
- ^ Ланска диджей (апрель 2000 г.). «Джордж Хантингтон (1850–1916) и наследственная хорея». Журнал истории нейронаук . 9 (1): 76–89. дои : 10.1076/0964-704X(200004)9:1;1-2;FT076 . ПМИД 11232352 . S2CID 22659368 .
- ^ Ослер В. (1908). «Исторические заметки о наследственной хорее» . Нейрографы . 1 (2): 113–116. Архивировано из оригинала 21 апреля 2022 года . Проверено 21 апреля 2022 г. См. стр. 115.
- См. также: Ослер В. (февраль 1893 г.). «Замечания о разновидностях хронической хореи и отчет о двух семьях наследственной формы с одним аутопсией» . Журнал нервных и психических заболеваний . 20 (2): 97–111. Архивировано из оригинала 21 апреля 2022 года . Проверено 21 апреля 2022 г. См. стр. 97.
- ^ Броуди И.А., Уилкинс Р.Х. (сентябрь 1967 г.). «Хорея Хантингтона». Архив неврологии . 17 (3): 331. doi : 10.1001/archneur.1967.00470270109013 . ПМИД 4228262 .
- ^ Бейтсон В. (1909). Принципы наследственности Менделя . Кембридж, Англия: Издательство Кембриджского университета. п. 229. Бейтсон называет «болезнь Хантингтона» «наследственной хореей».
- ^ Джеллифф С.Э., Манси Э.Б., Давенпорт CB (1913). «Хорея Хантингтона: исследование наследственности» . Журнал нервных и психических заболеваний . 40 (12): 796–799. дои : 10.1097/00005053-191312000-00010 . Архивировано из оригинала 5 апреля 2022 года . Проверено 19 сентября 2020 г.
- ^ Jump up to: а б Давенпорт CB, Манси EB (1916). «Хорея Хантингтона в связи с наследственностью и евгеникой» . Американский журнал безумия . 73 (2): 195–222. дои : 10.1176/ajp.73.2.195 .
- ^ Весси П.Р. (1932). «О передаче хореи Хантингтона за 300 лет – группа семьи Бурес» . Нервные и психические заболевания . 76 (6): 553–573. дои : 10.1097/00005053-193212000-00001 . S2CID 147656032 . Архивировано из оригинала 28 августа 2021 года . Проверено 1 апреля 2009 г.
- ^ Jump up to: а б Векслер А.Р. (2002). «Хорея и община в городе девятнадцатого века». Бюллетень истории медицины . 76 (3): 495–527. дои : 10.1353/bhm.2002.0150 . ПМИД 12486915 . S2CID 30791504 .
- ^ Коннели ПМ (май 1984 г.). «Болезнь Гентингтона: генетика и эпидемиология» . Американский журнал генетики человека . 36 (3): 506–26. ПМК 1684448 . ПМИД 6233902 .
- ^ Векслер Н.С. (2012). «Болезнь Хантингтона: пропаганда, движущая наука» . Ежегодный обзор медицины . 63 : 1–22. doi : 10.1146/annurev-med-050710-134457 . ПМИД 22248319 .
- ^ Jump up to: а б «Венесуэльский проект по болезни Хантингтона» . Сайт Фонда наследственных заболеваний . Фонд наследственных болезней. 2008. Архивировано из оригинала 10 августа 2015 года . Проверено 8 сентября 2008 г.
- ^ Jump up to: а б «Новый ген, содержащий тринуклеотидный повтор, который расширен и нестабильен на хромосомах при болезни Хантингтона. Совместная исследовательская группа по болезни Хантингтона». Клетка . 72 (6): 971–983. Март 1993 г. doi : 10.1016/0092-8674(93)90585-E . hdl : 2027.42/30901 . ПМИД 8458085 . S2CID 802885 .
- ^ Бертрам Л., Танзи Р.Э. (июнь 2005 г.). «Генетическая эпидемиология нейродегенеративных заболеваний» . Журнал клинических исследований . 115 (6): 1449–1457. дои : 10.1172/JCI24761 . ПМК 1137006 . ПМИД 15931380 .
- ^ Ла Спада А.Р., Ролинг Д.Б., Хардинг А.Е., Уорнер К.Л., Шпигель Р., Хаусманова-Петрусевич I и др. (декабрь 1992 г.). «Мейотическая стабильность и генотип-фенотипическая корреляция тринуклеотидного повтора при Х-сцепленной спинальной и бульбарной мышечной атрофии». Природная генетика . 2 (4): 301–4. дои : 10.1038/ng1292-301 . ПМИД 1303283 . S2CID 6603129 .
- ^ Jump up to: а б с Росс К.А., Тебризи С.Дж. (январь 2011 г.). «Болезнь Хантингтона: от молекулярного патогенеза к клиническому лечению». «Ланцет». Неврология . 10 (1): 83–98. дои : 10.1016/S1474-4422(10)70245-3 . ПМИД 21163446 . S2CID 17488174 .
- ^ Jump up to: а б Смит Дж. Э., Кордеро К. (23 мая 2023 г.). «Разыскал наука, а потом забыл» . Нью-Йорк Таймс . Архивировано из оригинала 23 мая 2023 года . Проверено 23 мая 2023 г.
- ^ «Что такое HD?» . Ассоциация болезни Гентингтона. Архивировано из оригинала 13 декабря 2011 года . Проверено 18 декабря 2011 г.
- ^ Давенпорт CB (май 1915 г.). «Хорея Хантингтона в связи с наследственностью и евгеникой» . Труды Национальной академии наук Соединенных Штатов Америки . 1 (5): 283–5. Бибкод : 1915PNAS....1..283D . дои : 10.1073/pnas.1.5.283 . ПМЦ 1090803 . ПМИД 16575999 .
- ^ Роллин Б.Е. (2006). «Регулирование исследований на животных и возникновение этики животных: концептуальная история» (PDF) . Теоретическая медицина и биоэтика . 27 (4): 285–304. дои : 10.1007/s11017-006-9007-8 . ПМИД 16937023 . S2CID 18620094 . Архивировано из оригинала (PDF) 8 октября 2020 года . Проверено 1 декабря 2019 г.
- ^ Дорфлингер Р.М. (февраль 2008 г.). «Проблема обмана в исследованиях эмбриональных стволовых клеток» . Пролиферация клеток . 41 (Приложение 1): 65–70. дои : 10.1111/j.1365-2184.2008.00492.x . ПМК 6496399 . ПМИД 18181947 .
- ^ Чепмен М.А. (июль 1990 г.). «Прогностическое тестирование генетических заболеваний, возникающих у взрослых: этические и юридические последствия использования анализа сцепления при болезни Хантингтона» . Американский журнал генетики человека . 47 (1): 1–3. ПМЦ 1683745 . ПМИД 2140926 .
- ^ Хаггинс М., Блох М., Канани С., Куоррел О.В., Тейлман Дж., Хедрик А. и др. (июль 1990 г.). «Этические и юридические дилеммы, возникающие во время прогностического тестирования заболеваний, начинающихся у взрослых: опыт болезни Хантингтона» . Американский журнал генетики человека . 47 (1): 4–12. ПМЦ 1683755 . ПМИД 1971997 .
- ^ «Мораторий на страхование генетики продлен до 2017 года» (Пресс-релиз). Ассоциация британских страховщиков. 5 апреля 2011 года. Архивировано из оригинала 4 марта 2016 года . Проверено 13 января 2016 г.
- ^ «Эксперт поддерживает раскрытие результатов генных тестов» . Статья Би-би-си. 7 июня 2007 г. Архивировано из оригинала 26 февраля 2008 г.
- ^ Бинеделл Дж., Солдан Дж.Р., Скурфилд Дж., Харпер П.С. (ноябрь 1996 г.). «Прогностическое тестирование болезни Хантингтона: аргументы в пользу подхода к оценке запросов подростков» . Журнал медицинской генетики . 33 (11): 912–8. дои : 10.1136/jmg.33.11.912 . ПМЦ 1050784 . ПМИД 8950670 .
- ^ Борри П., Гоффин Т., Найс Х., Дирикс К. (2008). «Прогностическое генетическое тестирование несовершеннолетних на генетические заболевания, возникающие во взрослом возрасте» . Медицинский журнал Mount Sinai, Нью-Йорк . 75 (3): 287–96. дои : 10.1002/msj.20038 . ПМИД 18704981 .
- ^ Jump up to: а б с Брауде П.Р., Де Верт ГМ, Эверс-Кибумс Дж., Петтигрю Р.А., Гераедтс Дж.П. (декабрь 1998 г.). «Преимплантационная генетическая диагностика болезни Хантингтона с неразглашением: практические и этические дилеммы». Пренатальная диагностика . 18 (13): 1422–6. doi : 10.1002/(SICI)1097-0223(199812)18:13<1422::AID-PD499>3.0.CO;2-R . ПМИД 9949442 . S2CID 39977672 .
- ^ «Международная ассоциация Хантингтона» . Международная ассоциация Хантингтона. 2013. Архивировано из оригинала 18 апреля 2009 года . Проверено 3 апреля 2009 г.
- ^ «Резолюция Сената США 531» . С. Рез. 531 . Сенат США. 6 апреля 2008 г. Архивировано из оригинала 17 ноября 2015 г. . Проверено 10 августа 2008 г.
- ^ Jump up to: а б с Одлинг-Сми Л. (17 мая 2007 г.). «Биомедицинская филантропия: денежное дерево» . Природа . 447 (7142): 251. Бибкод : 2007Natur.447..251. . дои : 10.1038/447251a . S2CID 4357517 .
- ^ Jump up to: а б «Болезнь Хантингтона: надежда через исследования» . www.ninds.nih.gov . Архивировано из оригинала 25 октября 2020 года . Проверено 16 ноября 2020 г.
- ^ Wild EJ, Тебризи SJ (сентябрь 2014 г.). «Цели будущих клинических исследований болезни Хантингтона: что находится в стадии разработки?» . Двигательные расстройства . 29 (11): 1434–45. дои : 10.1002/mds.26007 . ПМК 4265300 . ПМИД 25155142 .
- ^ Jump up to: а б Соломон С. (3 мая 2017 г.). «Таубе выделит 3 миллиона долларов на исследования болезни Хантингтона в США» . Таймс Израиля . Архивировано из оригинала 3 мая 2017 года . Проверено 5 мая 2017 г.
- ^ «Научные публикации | Фонд CHDI» . chdifoundation.org . Архивировано из оригинала 12 декабря 2021 года . Проверено 12 декабря 2021 г.
- ^ «Фонд ЧДИ» . chdifoundation.org . Архивировано из оригинала 14 ноября 2020 года . Проверено 13 ноября 2020 г.
- ^ Проверьте E (май 2007 г.). «Биомедицинская филантропия: любовь или деньги» . Природа . 447 (7142): 252–3. Бибкод : 2007Natur.447..252C . дои : 10.1038/447252a . ПМИД 17507955 . S2CID 4318384 .
- ^ Jump up to: а б Муньос-Санджуан I, врач Бейтса (февраль 2011 г.). «Важность интеграции фундаментальных и клинических исследований для разработки новых методов лечения болезни Хантингтона» . Журнал клинических исследований . 121 (2): 476–83. дои : 10.1172/JCI45364 . ПМК 3026740 . ПМИД 21285520 .
- ^ МакБрайд Дж.Л., Питцер М.Р., Будро Р.Л., Дюфур Б., Хоббс Т., Охеда С.Р. и др. (декабрь 2011 г.). «Доклиническая безопасность РНКи-опосредованного подавления HTT у макак-резус как потенциального лечения болезни Хантингтона» . Молекулярная терапия . 19 (12): 2152–62. дои : 10.1038/mt.2011.219 . ПМЦ 3242667 . ПМИД 22031240 .
- ^ Кордасевич Х.Б., Станек Л.М., Ванцевич Е.В., Мазур С., Макалонис М.М., Пытел К.А. и др. (июнь 2012 г.). «Устойчивое терапевтическое обращение болезни Хантингтона за счет временного подавления синтеза хантингтина» . Нейрон . 74 (6): 1031–44. дои : 10.1016/j.neuron.2012.05.009 . ПМЦ 3383626 . ПМИД 22726834 .
- ^ Барнс Д.В., Уитли Р.Дж. (февраль 1987 г.). «Противовирусная терапия и легочные заболевания». Грудь . 91 (2): 246–51. дои : 10.1378/сундук.91.2.246 . ПМИД 3026739 .
- ^ «Начинается суд над Лэндмарком Хантингтоном» . Архивировано из оригинала 21 октября 2015 года . Проверено 19 октября 2015 г.
- ^ «Безопасность, переносимость, фармакокинетика и фармакодинамика IONIS-HTTRx у пациентов с ранней манифестацией болезни Хантингтона - полный текст» . ClinicalTrials.gov. Архивировано из оригинала 29 сентября 2015 года . Проверено 18 апреля 2016 г.
- ^ Уайлд Э.Дж., Богджио Р., Лангбен Д., Робертсон Н., Хайдер С., Миллер Дж.Р. и др. (май 2015 г.). «Количественное определение мутантного белка хантингтина в спинномозговой жидкости пациентов с болезнью Гентингтона» . Журнал клинических исследований . 125 (5): 1979–86. дои : 10.1172/jci80743 . ПМЦ 4463213 . ПМИД 25844897 .
- ^ Чейз А (май 2015 г.). «Болезнь Хантингтона: спинномозговая жидкость и биомаркеры МРТ для продромальной HD». Обзоры природы Неврология . 11 (5): 245. doi : 10.1038/nrneurol.2015.63 . ПМИД 25896083 . S2CID 38300571 .
- ^ Кейзер М.С., Кордасевич Х.Б., Макбрайд Дж.Л. (апрель 2016 г.). «Стратегии подавления генов при доминантно наследственных нейродегенеративных заболеваниях: уроки болезни Хантингтона и спиноцеребеллярной атаксии» . Молекулярная генетика человека . 25 (Р1): Р53–64. дои : 10.1093/hmg/ddv442 . ПМЦ 4802374 . ПМИД 26503961 .
- ^ Квон Д. (6 апреля 2021 г.). «Генетическая терапия дает новую надежду на борьбу с неизлечимыми заболеваниями головного мозга». Природа . 592 (7853): 180–183. Бибкод : 2021Natur.592..180K . дои : 10.1038/d41586-021-00870-x . ПМИД 33824521 . S2CID 233173862 .
- ^ Хардинг Р. (26 апреля 2021 г.). Фокс Л. (ред.). «Обзор клинических исследований болезни Хантингтона» . HDBuzz . Архивировано из оригинала 5 мая 2021 года . Проверено 5 мая 2021 г.
- ^ Jump up to: а б Джаджадикерта А., Кешри С., Павел М., Престил Р., Райан Л., Рубинштейн, округ Колумбия (апрель 2020 г.). «Индукция аутофагии как терапевтическая стратегия нейродегенеративных заболеваний» . Журнал молекулярной биологии . Аутофагия при нейродегенеративных заболеваниях. 432 (8): 2799–2821. дои : 10.1016/j.jmb.2019.12.035 . ПМИД 31887286 . S2CID 209518157 . Архивировано из оригинала 23 мая 2022 года . Проверено 20 июня 2023 г.
- ^ Равикумар Б., Вашер С., Бергер З., Дэвис Дж.Э., Луо С., Ороз Л.Г. и др. (июнь 2004 г.). «Ингибирование mTOR вызывает аутофагию и снижает токсичность экспансии полиглутамина на моделях болезни Хантингтона на мухах и мышах» . Природная генетика . 36 (6): 585–595. дои : 10.1038/ng1362 . ПМИД 15146184 . S2CID 7749825 .
- ^ Холли С.М., Камджу Т., Рейдлинг Дж.К., Фьюри Б., Коул-Бергум Д., Бауэр Г. и др. (апрель 2018 г.). «Терапевтические эффекты стволовых клеток на моделях болезни Хантингтона на грызунах: обзор и электрофизиологические данные» . Нейронауки и терапия ЦНС . 24 (4). Уайли: 329–342. дои : 10.1111/cns.12839 . ПМК 6489814 . ПМИД 29512295 .
- ^ Кандифф П.Е., Андерсон С.А. (июнь 2011 г.). «Влияние индуцированных плюрипотентных стволовых клеток на изучение заболеваний центральной нервной системы» . Текущее мнение в области генетики и развития . 21 (3): 354–61. дои : 10.1016/j.где.2011.01.008 . ПМЦ 3932563 . ПМИД 21277194 .
- ^ Jump up to: а б Стоквелл Б.Р., Фридман Анджели Дж.П., Байир Х., Буш А.И., Конрад М., Диксон С.Дж. и др. (октябрь 2017 г.). «Ферроптоз: регулируемая связь клеточной смерти, связывающая метаболизм, окислительно-восстановительную биологию и болезни» . Клетка . 171 (2): 273–285. дои : 10.1016/j.cell.2017.09.021 . ПМЦ 5685180 . ПМИД 28985560 .
- ^ «Поиск: Болезнь Хантингтона — Результаты списка — ClinicalTrials.gov» . www.clinicaltrials.gov . Архивировано из оригинала 11 ноября 2021 года . Проверено 16 ноября 2020 г.
- ^ «Завершенные клинические испытания» . Исследовательская группа Хантингтона. Архивировано из оригинала 28 июня 2012 года . Проверено 4 февраля 2012 г.
Внешние ссылки
[ редактировать ]- Болезнь Хантингтона у Керли
- Проект HOPES. Архивировано 27 августа 2020 года в Wayback Machine - информационный проект Стэнфордского университета в формате HD.
- HDBuzz – новости исследований HD, написанные учеными простым языком
- HD Drug Works – новости о текущих методах лечения и запланированных исследованиях
- болезнь Хантингтона
- Аутосомно-доминантные заболевания
- Расстройства, вызывающие судороги
- Болезни, названные в честь первооткрывателей
- Экстрапирамидные и двигательные расстройства
- Генетические заболевания и расстройства
- Системные атрофии, поражающие преимущественно центральную нервную систему.
- Нарушения тринуклеотидных повторов