Альдольная реакция
| Альдольное дополнение | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Тип реакции | Реакция сцепления | ||||||||
| Реакция | |||||||||
| |||||||||
| Условия | |||||||||
| Температура | -Д, ~-70°С [ а ]
| ||||||||
| Катализатор | - ОН или Ч +
| ||||||||
| Идентификаторы | |||||||||
| Портал органической химии | альдольное присоединение | ||||||||
| RSC Идентификатор онтологии | RXNO: 0000016 | ||||||||
Альдольная реакция ( альдольное присоединение ) — это реакция в органической химии , которая объединяет два карбонильных соединения (например, альдегиды или кетоны ) с образованием нового β-гидроксикарбонильного соединения. форма может включать нуклеофильное присоединение енолированного Его простейшая кетона к другому:

Эти продукты известны как альдолы , от сочетания альдегид + спирт — структурного мотива, который можно увидеть во многих продуктах. Использование альдегида в названии связано с его историей: альдегиды более реакционноспособны, чем кетоны, поэтому реакция с ними была открыта первой. [ 2 ] [ 3 ] [ 4 ]
Альдольная реакция является парадигмой органической химии и, возможно, наиболее распространенным способом образования углерод-углеродных связей в органической химии . [ 5 ] [ 6 ] [ 7 ] Он дал свое название семейству альдольных реакций , и аналогичные методы анализа целого семейства реакций карбонильного α-замещения , а также конденсации дикетона . Когда нуклеофил и электрофил различны, реакция называется скрещенной альдольной реакцией ; и наоборот, когда нуклеофил и электрофил одинаковы, реакция называется альдольной димеризацией .
Альдольные структурные единицы обнаружены во многих важных молекулах, как природных, так и синтетических. [ 8 ] [ 9 ] Реакция используется в нескольких промышленных синтезах, в частности пентаэритрита . [ 10 ] триметилолпропан , пластификатор 2-этилгексанол и препарат Липитор ( аторвастатин , кальциевая соль). [ 11 ] Для многих промышленных применений стереохимия альдольной реакции не важна, но эта тема представляет большой интерес для синтеза многих специальных химических веществ.

Колба справа представляет собой раствор диизопропиламида лития (ЛДА) в тетрагидрофуране (ТГФ). В колбе слева находится раствор енолята лития трет -бутилпропионата (образующегося при добавлении LDA к трет -бутилпропионату). Затем в енолятную колбу можно добавить альдегид, чтобы инициировать реакцию альдольного присоединения.
Обе колбы погружают в охлаждающую баню из сухого льда и ацетона (-78 °C), температуру которой контролируют с помощью термопары (провод слева).
Механизмы
[ редактировать ]Альдольная реакция имеет один основной механизм, но она проявляется в разных формах в зависимости от pH: [ 12 ]
Если катализатором является умеренное основание, такое как гидроксид -ион или алкоксид , альдольная реакция происходит в результате нуклеофильной атаки резонансно -стабилизированного енолята на карбонильную группу другой молекулы. Продукт представляет собой алкоксидную соль альдольного продукта. Затем образуется сам альдоль, который затем может подвергнуться дегидратации с образованием ненасыщенного карбонильного соединения. На схеме показан простой механизм катализируемой основаниями альдольной реакции альдегида с самим собой.

Хотя в некоторых случаях требуется только каталитическое количество основания, более обычной процедурой является использование стехиометрического количества сильного основания, такого как LDA или NaHMDS . В этом случае образование енолята является необратимым, и альдольный продукт не образуется до тех пор, пока алкоксид металла альдольного продукта не будет протонирован на отдельной стадии обработки.
При использовании кислотного катализатора начальная стадия механизма реакции включает катализируемую кислотой таутомеризацию карбонильного соединения в енол. Кислота также служит для активации карбонильной группы другой молекулы путем протонирования, что делает ее очень электрофильной. Енол является нуклеофильным по отношению к α-углероду, что позволяет ему атаковать протонированное карбонильное соединение, что приводит к образованию альдола после депротонирования . Некоторые из них могут также дегидратироваться помимо целевого продукта с образованием ненасыщенного карбонильного соединения посредством альдольной конденсации .

Контроль реагента скрещенных альдолов
[ редактировать ]Несмотря на привлекательность альдольного многообразия, существует несколько проблем, которые необходимо решить, чтобы сделать процесс эффективным. Первая проблема носит термодинамический характер: большинство альдольных реакций обратимы. Кроме того, в случае простых альдегид-кетон-альдольных реакций равновесие также едва ли находится на стороне продуктов. [ 13 ] Если условия особенно суровые (например: NaOMe/MeOH/ рефлюкс ), может возникнуть конденсация, но ее обычно можно избежать с помощью мягких реагентов и низких температур (например, LDA (сильное основание), ТГФ, -78 °C). Хотя присоединение альдолов обычно происходит почти до завершения в необратимых условиях, изолированные альдольные аддукты чувствительны к индуцированному основанием расщеплению ретро-альдолов с возвратом исходных материалов. Напротив, ретро-альдольные конденсации редки, но возможны. [ 14 ] На этом основана каталитическая стратегия альдолаз I класса в природе, а также многочисленных низкомолекулярных аминных катализаторов. [ 15 ]
При реакции смеси несимметричных кетонов скрещенных альдолов ( присоединения ): можно ожидать четыре продукта

Таким образом, если кто-то желает получить только один из перекрестных продуктов, необходимо контролировать, какой карбонил становится нуклеофильным енолом/енолятом, а какой остается в своей электрофильной карбонильной форме. Самый простой контроль – это если только один из реагентов имеет кислые протоны и только эта молекула образует енолят. Например, при добавлении диэтилмалоната к бензальдегиду образуется только один продукт:

Если одна группа значительно более кислая, чем другая, наиболее кислый протон отрывается основанием, и на этом карбониле образуется енолят, в то время как менее кислый карбонил остается электрофильным. Этот тип контроля работает только в том случае, если разница в кислотности достаточно велика и основание является ограничивающим реагентом . Типичным субстратом для этой ситуации является ситуация, когда депротонируемое положение активируется более чем одной карбонильноподобной группой. Общие примеры включают группу CH 2 , фланкированную двумя карбонилами или нитрилами (см., например, конденсацию Кнёвенагеля и первые стадии синтеза малонового эфира и синтеза ацетоуксусного эфира ).
В противном случае наиболее кислые карбонилы обычно являются также и наиболее активными электрофилами: сначала альдегиды , затем кетоны , затем сложные эфиры и, наконец, амиды . Таким образом, реакции с перекрестными альдегидами обычно являются наиболее сложными, поскольку они могут легко полимеризоваться или реагировать неселективно, образуя статистическую смесь продуктов. [ 16 ]
Одним из распространенных решений является сначала образование енолята одного партнера, а затем добавление другого партнера под кинетическим контролем . [ 17 ] Кинетический контроль означает, что прямая реакция альдольного присоединения должна протекать значительно быстрее, чем обратная ретро-альдольная реакция. Для успеха этого подхода необходимо также выполнить два других условия; должна быть возможность количественно образовать енолят одного партнера, и прямая альдольная реакция должна быть значительно быстрее, чем передача енолята от одного партнера к другому. Обычные условия кинетического контроля включают образование енолята кетона с LDA при -78 ° C с последующим медленным добавлением альдегида.
Стереоселективность
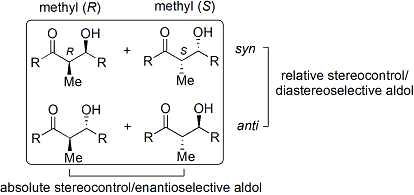
[ редактировать ]Альдольная реакция объединяет две относительно простые молекулы в более сложную. Повышенная сложность возникает из-за того, что каждый конец новой связи может стать стереоцентром . Современная методология не только разработала высокопродуктивные альдольные реакции, но и полностью контролирует как относительную, так и абсолютную конфигурацию этих новых стереоцентров. [ 6 ]
химии сахаридов эритро/трео Для описания относительной стереохимии α- и β-углеродов в старых статьях используется номенклатура ; более современные статьи используют следующее соглашение син / анти . Когда пропионатные нуклеофилы (или нуклеофилы более высокого порядка) присоединяются к альдегидам, читатель визуализирует группу R кетона и группу R' альдегида, выстроенных в виде «зигзага» на бумаге (или экране). Расположение образовавшихся стереоцентров считается син- или анти -в зависимости от того, находятся ли они по одну или противоположные стороны основной цепи:
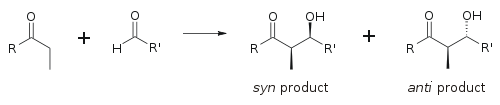
альдольной реакции, Основным фактором, определяющим стереоселективность является енолизирующий противоион металла . Более короткие связи металл-кислород «затягивают» переходное состояние и вызывают большую стереоселекцию. [ 18 ] Бор часто используют [ 19 ] [ 20 ] потому что длина его связей значительно короче, чем у других дешевых металлов ( лития , алюминия или магния ). Следующая реакция дает соотношение син:анти 80:20 при использовании енолята лития по сравнению с 97:3 при использовании енолята бибутилбора.

Если противоион определяет силу стереоиндукции , то енолятный изомер определяет ее направление . E Изомеры дают антипродукты , а Z - син : [ 21 ]


Модель Циммермана-Тракслера
[ редактировать ]Если два реагента имеют карбонилы, соседние с ранее существовавшим стереоцентром, то новые стереоцентры могут образовываться с фиксированной ориентацией относительно старого . Этот «стереоконтроль на основе субстрата» широко изучался, и примеры изобилуют литературой. Во многих случаях стилизованное переходное состояние , называемое моделью Циммермана-Тракслера , может предсказать новую ориентацию на основе конфигурации 6-членного кольца . [ 22 ]
Об еноле
[ редактировать ]Если енол имеет соседний стереоцентр, то два стереоцентра, фланкирующих карбонил в продукте, естественным образом являются син : [ 23 ]

Основная механистическая причина зависит от изомера енола. Для енолята E стереоиндукция необходима, чтобы избежать 1,3- аллильного напряжения , тогда как енолят Z вместо этого стремится избежать 1,3- диаксиальных взаимодействий : [ 24 ]

Однако Fráter & Seebach показали , что хелатирующая основная группа Льюиса, примыкающая к енолу, вместо этого будет вызывать антиприсоединение .
На электрофиле
[ редактировать ]Еноляты E демонстрируют диастереофасную селекцию Фелкина , тогда как еноляты Z проявляют селективность против Фелкина. Общая модель представлена ниже: [ 25 ] [ 26 ]
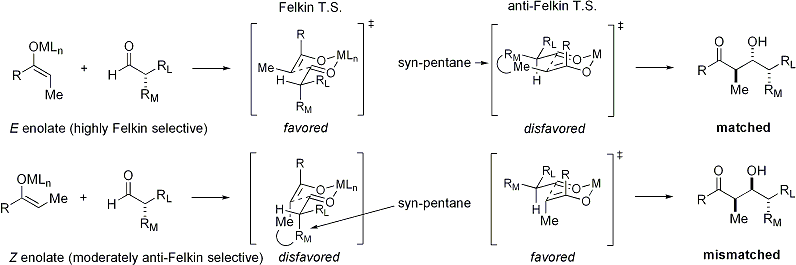
Поскольку переходное состояние для Z -енолятов должно содержать либо дестабилизирующее син -пентановое взаимодействие, либо анти-Фелькин -ротамер , Z -еноляты менее диастереоселективны: [ 27 ] [ 28 ]

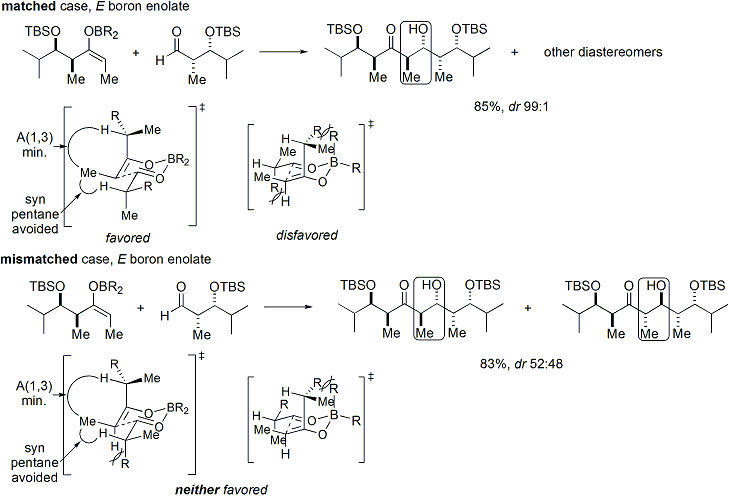
На обоих
[ редактировать ]Если и енолят, и альдегид содержат уже существующую хиральность, то результат альдольной реакции «двойного стереодифференцирования» можно предсказать с использованием объединенной стереохимической модели, которая учитывает все эффекты, обсуждавшиеся выше. [ 29 ] Вот несколько примеров: [ 28 ]
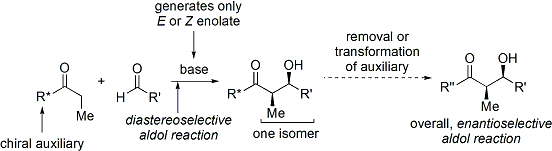
Хиральные вспомогательные вещества оксазолидинона
[ редактировать ]В конце 1970-х и 1980-х годах Дэвид А. Эванс и его коллеги разработали метод стереоселекции при альдольном синтезе альдегидов и карбоновых кислот . [ 30 ] [ 31 ] Этот метод работает путем временного добавления хирального оксазолидинона вспомогательного для создания хирального енолята. Ранее существовавшая хиральность вспомогательного вещества затем передается альдольному аддукту с помощью методов Циммермана-Тракслера, а затем оксазолидинон отщепляется.
Коммерческие оксазолидиноны относительно дороги, но их получают в два этапа синтеза из сравнительно недорогих аминокислот. (Экономичные крупномасштабные синтезы готовят вспомогательные вещества собственными силами.) Во-первых, боргидрид восстанавливает кислотную часть . Затем полученный аминоспирт дегидратационно циклизуется с простым эфиром карбоната, например диэтилкарбонатом.
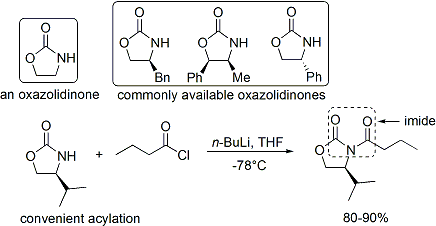
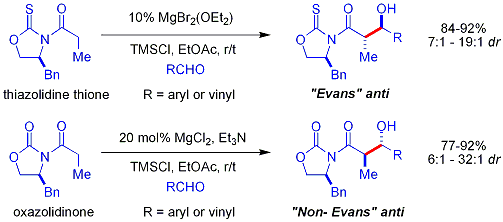
Ацилирование . оксазолидинона неофициально называют «загрузкой»
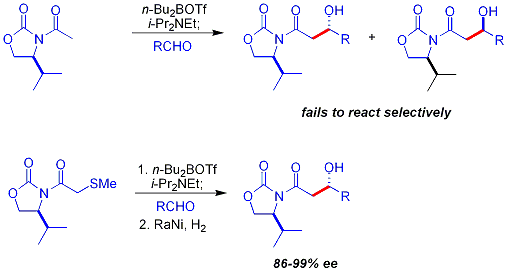
Антиаддукты , для которых требуется енолят Е , невозможно надежно получить методом Эванса. Однако еноляты Z , приводящие к синаддуктам , можно надежно получить с помощью бор-опосредованной мягкой енолизации: [ 32 ]

Часто один диастереомер можно получить путем одной кристаллизации альдольного аддукта.
Многие методы отщепляют вспомогательные: [ 33 ]

Вариации
[ редактировать ]Обычным дополнительным хиральным вспомогательным веществом является тиоэфирная группа: [ 33 ] [ б ]
Кримминс тиазолидинетион альдол
[ редактировать ]В тиазолидинетионовом подходе Кримминса: [ 34 ] [ 35 ] тиазолидинетион является хиральным вспомогательным веществом [ 36 ] и может производить аддукты «син-Эванса» или «син-не-Эванса», просто варьируя количество (-)-спартеина . Считается, что реакция протекает через шестичленные переходные состояния , связанные с титаном , аналогичные предполагаемым переходным состояниям для вспомогательного вещества Эванса.

«Маскированные» енолы
[ редактировать ]В обычной модификации альдольной реакции используются другие функциональные группы, аналогичные эрзац- енолам. В альдольной реакции Мукаямы [ 37 ] Эфиры силиленола присоединяются к карбонилам в присутствии катализатора кислоты Льюиса , такого как трифторид бора (в виде эфирата трифторида бора ) или тетрахлорид титана . [ 38 ] [ 39 ]
При енаминовом алкилировании по Сторку вторичные амины образуют енамины при воздействии кетонов . Эти енамины затем реагируют (возможно, энантиоселективно) [ 40 ] ) с подходящими электрофилами. Эта стратегия предлагает простой энантиоселектив без переходных металлов. В отличие от предпочтения синаддуктов , которое обычно наблюдается при присоединении альдолов на основе енолята, эти добавления альдолов являются антиселективными .
В водном растворе енамин затем может быть гидролизован из продукта, что делает его катализатором на основе небольших органических молекул . Яркий пример: пролин эффективно катализирует циклизацию трикетона:
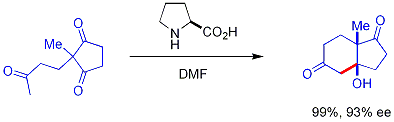
Эта комбинация представляет собой реакцию Хаджоса-Пэрриша. [ 41 ] [ 42 ] [ 43 ] В условиях Хайоса-Пэрриша необходимо только каталитическое количество пролина (3 мол.%). Опасности ахиральной фоновой реакции нет, поскольку временные промежуточные енаминовые соединения гораздо более нуклеофильны, чем их исходные кетоненолы.
Стратегия типа аиста также допускает сложные перекрестные реакции между двумя альдегидами. Во многих случаях условия достаточно мягкие, чтобы избежать полимеризации: [ 44 ]
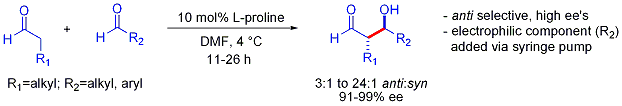
Однако селективность требует медленного добавления желаемого электрофильного партнера, контролируемого шприцевым насосом, поскольку оба реагирующих партнера обычно имеют енолизируемые протоны. Если один альдегид не имеет енолизируемых протонов или альфа- или бета-разветвлений, можно достичь дополнительного контроля.
«Прямое» альдольное присоединение
[ редактировать ]При обычном альдольном присоединении карбонильное соединение депротонируется с образованием енолята. Енолят добавляют к альдегиду или кетону, который образует алкоксид, который затем протонируется при обработке. В принципе, лучший метод позволил бы избежать необходимости многостадийной последовательности в пользу «прямой» реакции, которую можно было бы провести за одну стадию процесса.
Если один из партнеров сочетания предпочтительно енолизируется, то общая проблема заключается в том, что при добавлении образуется алкоксид, который является гораздо более основным, чем исходные материалы. Этот продукт прочно связывается с енолизирующим агентом, не позволяя ему катализировать дополнительные реагенты:
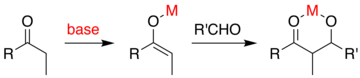
Один из подходов, продемонстрированный Эвансом, заключается в силилировании альдольного аддукта. [ 45 ] [ 46 ] кремниевый реагент, такой как TMSCl В реакцию добавляется , который заменяет металл на алкоксид, обеспечивая обмен металлического катализатора:
Использование в синтезе углеводов
[ редактировать ]В традиционных синтезах гексоз используются вариации итеративных стратегий защиты-снятия защиты , требующие 8–14 этапов. Органокатализ может получить доступ ко многим из одних и тех же субстратов с помощью двухэтапного протокола, включающего катализируемую пролином димеризацию альфа-оксиальдегидов с последующей тандемной альдольной циклизацией Мукаямы.
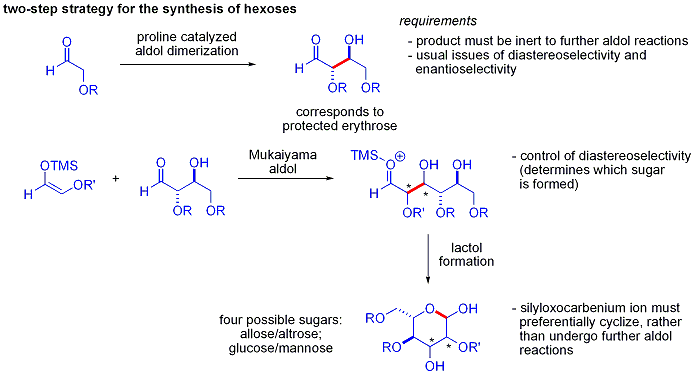
Альдольная димеризация альфа-оксиальдегидов требует, чтобы альдольный аддукт, который сам является альдегидом, был инертным к дальнейшим альдольным реакциям. [ 47 ] Более ранние исследования показали, что альдегиды, содержащие альфа-алкилокси- или альфа- силилокси -заместители, подходят для этой реакции, тогда как альдегиды, содержащие электроноакцепторные группы, такие как ацетокси, нереакционноспособны. Защищенный продукт эритрозы затем можно было превратить в четыре возможных сахара путем добавления альдолов Мукаямы с последующим образованием лактола . Это требует соответствующего диастереоконтроля при присоединении альдолов Мукаямы и образовавшегося иона силилоксикарбения для преимущественной циклизации, а не для дальнейшей альдольной реакции. В итоге глюкоза , манноза и аллоза были синтезированы :


Биологические альдольные реакции
[ редактировать ]Примеры альдольных реакций в биохимии включают расщепление фруктозо-1,6-бисфосфата на дигидроксиацетон и глицеральдегид-3-фосфат на четвертой стадии гликолиза , что является примером обратной («ретро») альдольной реакции, катализируемой ферментом. альдолаза А (также известная как фруктозо-1,6-бисфосфатальдолаза).
В глиоксилатном цикле растений и некоторых прокариот изоцитратлиаза образует глиоксилат и сукцинат из изоцитрата . После депротонирования ОН-группы изоцитратлиаза расщепляет изоцитрат на четырехуглеродный сукцинат и двухуглеродный глиоксилат посредством реакции альдольного расщепления. Это расщепление по механизму аналогично альдолазной реакции гликолиза.
История
[ редактировать ]Альдольная реакция была независимо открыта русским химиком (и композитором-романтиком) Александром Бородиным в 1869 году. [ 48 ] [ 49 ] [ 50 ] и французским химиком Шарлем-Адольфом Вюрцем в 1872 году, который первоначально использовал для проведения реакции альдегиды. [ 2 ] [ 3 ] [ 4 ]
Говард Циммерман и Марджори Д. Трэкслер предложили свою модель стереоиндукции в статье 1957 года. [ 22 ]
См. также
[ редактировать ]- Реакция Альдоля-Тищенко
- Реакция Бейлиса-Хиллмана
- Реакция Иванова
- Реформатская реакция
- Конденсация Кляйзена-Шмидта
Примечания
[ редактировать ]- ^ Обычно лучше минимизировать нагревание для этой реакции. Поскольку удаление воды из-за избыточного тепла рискует сместить равновесие в пользу реакции дегидратации, приводящей к образованию продукта альдольной конденсации.
Избегая нагревания, можно избежать обезвоживания, так что большая часть производимого продукта представляет собой продукт присоединения альдолов. [ 1 ] - ^ В этой реакции нуклеофилом является енолят бора, полученный в результате реакции с трифлатом дибутилбора (nBu 2 BOTf), основанием является N,N-диизопропилэтиламин . Тиоэфир удаляется на этапе 2 путем никеля Ренея / водородного восстановления.
Ссылки
[ редактировать ]- ^ Кляйн, Дэвид Р. (22 декабря 2020 г.). Органическая химия (4-е изд.). Хобокен, Нью-Джерси: Уайли. п. 1014. ИСБН 978-1-119-65959-4 . OCLC 1201694230 .
- ^ Jump up to: а б Вюрц, Калифорния (1872 г.). «Об альдегидном спирте» . Бюллетень Парижского химического общества . 2-я серия (на французском языке). 17 : 436–442.
- ^ Jump up to: а б Вюрц, Калифорния (1872 г.). «Об альдегидном спирте» . Журнал практической химии (на немецком языке). 5 (1): 457–464. дои : 10.1002/prac.18720050148 .
- ^ Jump up to: а б Вюрц, Калифорния (1872 г.). «Sur un aldehyde-alcool» [Об альдегидном спирте]. Доклады Академии наук (на французском языке). 74 : 1361.
- ^ Уэйд, LG (2005). Органическая химия (6-е изд.). Река Аппер-Сэддл, Нью-Джерси: Прентис-Холл. стр. 1056–66. ISBN 978-0-13-236731-8 .
- ^ Jump up to: а б Смит, Майкл Б.; Марш, Джерри (2006). Продвинутая органическая химия марта: реакции, механизмы и структура . дои : 10.1002/0470084960 . ISBN 9780470084960 .
- ^ Марвальд, Р. (2004). Современные альдольные реакции, тома 1 и 2 . Вайнхайм, Германия: Wiley-VCH Verlag GmbH & Co. KGaA. стр. 1218–23 . ISBN 978-3-527-30714-2 .
- ^ Хиткок, Швейцария (1991). «Альдольная реакция: кислотный и основной катализ». Трост , BM ; Флеминг, И. (ред.). Комплексный органический синтез . Том. 2. Эльзевир Наука. стр. 133–179. дои : 10.1016/B978-0-08-052349-1.00027-5 . ISBN 978-0-08-052349-1 .
- ^ Патерсон, И. (1988). «Новая методология асимметричного альдола с использованием енолятов бора». хим. Индий . 12 : 390–394.
- ^ Местрес Р. (2004). «Зеленый взгляд на альдольную реакцию». Зеленая химия . 6 (12): 583–603. дои : 10.1039/b409143b .
- ^ Цзе Джек Ли; и др. (2004). Современный синтез лекарств . Уайли-Интерсайенс. стр. 118–. ISBN 978-0-471-21480-9 .
- ^ Гроссманн, Роберт Б. (январь 2002 г.). Искусство написания разумных механизмов органических реакций (2-е изд.). Нью-Йорк: Спрингер. п. 133. ИСБН 0-387-95468-6 .
- ^ Моландер, Джорджия, изд. (2011). Стереоселективный синтез 2: Стереоселективные реакции карбонильных и иминогрупп (1-е изд.). Штутгарт: Георг Тиме Верлаг. дои : 10.1055/sos-sd-202-00331 . ISBN 978-3-13-154121-5 .
- ^ Гатри, JP; Купер, К.Дж.; Коссар, Дж.; Доусон, бакалавр; Тейлор, К.Ф. (1984). «Ретроальдольная реакция коричного альдегида» . Может. Дж. Хим. 62 (8): 1441–1445. дои : 10.1139/v84-243 .
- ^ Моландер, изд. (2011). Стереоселективный синтез 2: Стереоселективные реакции карбонильных и иминогрупп (1-е изд.). Штутгарт: Георг Тиме Верлаг. дои : 10.1055/sos-sd-202-00331 . ISBN 978-3-13-154121-5 .
- ^ Уоррен, Стюарт; Вятт, Пол (2008). Органический синтез: подход отключения (2-е изд.). Уайли. ISBN 978-0-470-71236-8 .
- ^ Бал, Б.; Автобус, Коннектикут; Смит, К.; Хиткок, CH, (2SR,3RS)-2,4-диметил-3-гидроксипентановая кислота. Архивировано 6 июня 2011 г. в Wayback Machine , Org. Синтез. , Колл. Том. 7, с.185 (1990); Том. 63, с.89 (1985).
- ^ Эванс, Пенсильвания ; Нельсон СП; Фогель Э.; Табер Т.Р. (1981). «Стереоселективные альдольные конденсации через еноляты бора». Журнал Американского химического общества . 103 (11): 3099–3111. дои : 10.1021/ja00401a031 .
- ^ Кауден, CJ; Патерсон, И. Орг. Реагировать. 1997 , 51 , 1.
- ^ Коуден, CJ; Патерсон, И. (2004). Асимметричные альдольные реакции с использованием енолятов бора . Органические реакции. стр. 1–200. дои : 10.1002/0471264180.или051.01 . ISBN 978-0471264187 .
- ^ Браун, ХК ; Дхар, РК; Бакши, РК; Пандиараджан, ПК; Сингарам, Б. (1989). «Основное влияние уходящей группы в хлоридах и трифлатах диалкилбора на контроль стереоспецифического превращения кетонов в боринаты E- или Z-енола». Журнал Американского химического общества . 111 (9): 3441–3442. дои : 10.1021/ja00191a058 .
- ^ Jump up to: а б Циммерман, HE; Тракслер, доктор медицины (1957). «Стереохимия реакций Иванова и Реформатского. I». Журнал Американского химического общества . 79 (8): 1920–1923. дои : 10.1021/ja01565a041 .
- ^ Эванс, Пенсильвания ; Ригер Д.Л.; Билодо МТ; Урпи Ф. (1991). «Стереоселективные альдольные реакции енолятов хлортитана. Эффективный метод сборки синтонов, связанных с полипропионатом». Журнал Американского химического общества . 113 (3): 1047–1049. дои : 10.1021/ja00003a051 .
- ^ Хиткок, Швейцария ; Автобус, Коннектикут; Клешник, Вашингтон; Пиррунг, MC; Зон, Дж. Э.; Лампе, Дж. (1980). «Ациклическая стереоселекция. 7. Стереоселективный синтез 2-алкил-3-гидроксикарбонильных соединений альдольной конденсацией». Журнал органической химии . 45 (6): 1066–1081. дои : 10.1021/jo01294a030 .
- ^ Эванс, Д.А.; Нельсон, СП; Табер, Т.Р. (1982). «Стереоселективные альдольные конденсации». Темы стереохимии . Том. 13. С. 1–115. ISBN 9780471056805 .
- ^ Руш В.Р. (1991). «Относительно диастереофациальной селективности альдольных реакций альфа-метилхиральных альдегидов и енолятов пропионатов лития и бора». Журнал органической химии . 56 (13): 4151–4157. дои : 10.1021/jo00013a015 .
- ^ Масамунэ С.; Эллингбо Дж.В.; Чой В. (1982). «Альдольная стратегия: координация катиона лития с алкокси-заместителем». Журнал Американского химического общества . 104 (20): 1047–1049. дои : 10.1021/ja00384a062 .
- ^ Jump up to: а б Эванс, Пенсильвания ; Дарт М.Дж.; Даффи Дж.Л.; Ригер Д.Л. (1995). «Двойные стереодифференцирующие альдольные реакции. Документирование «частично совпадающих» конструкций альдольных связей в сборке полипропионатных систем». Журнал Американского химического общества . 117 (35): 9073–9074. дои : 10.1021/ja00140a027 .
- ^ Масамунэ С.; Чой В.; Петерсен Дж.С.; Сита ЛР (1985). «Двойной асимметричный синтез и новая стратегия стереохимического контроля в органическом синтезе». Энджью. хим. Межд. Эд. англ. 24 : 1–30. дои : 10.1002/anie.198500013 .
- ^ Эванс, Д.А. (1982). «Исследования в области асимметричного синтеза: разработка практических хиральных енолят-синтонов» (PDF) . Альдрихимика Акта . 15:23 .
- ^ Гейдж-младший; Эванс Д.А., Диастереоселективная альдольная конденсация с использованием хирального вспомогательного вещества оксазолидинона: (2S*,3S*)-3-гидрокси-3-фенил-2-метилпропановая кислота. Архивировано 29 сентября 2012 г. в Wayback Machine , Organic Syntheses , Coll. Том. 8, с.339 (1993); Том. 68, с.83 (1990).
- ^ Эванс, Пенсильвания ; Бартроли Дж.; Ши ТЛ (1981). «Энантиоселективные альдольные конденсации. 2. Эритроселективные хиральные альдольные конденсации через еноляты бора». Журнал Американского химического общества . 103 (8): 2127–2129. дои : 10.1021/ja00398a058 .
- ^ Jump up to: а б Эванс, Пенсильвания ; Бендер С.Л.; Моррис Дж. (1988). «Полный синтез полиэфирного антибиотика Х-206». Журнал Американского химического общества . 110 (8): 2506–2526. дои : 10.1021/ja00216a026 .
- ^ Кримминс МТ; Кинг Б.В.; Табет А.Е. (1997). «Асимметричные альдольные присоединения к енолятам титана ацилоксазолидинтионов: зависимость селективности от аминного основания и стехиометрии кислоты Льюиса». Журнал Американского химического общества . 119 (33): 7883–7884. дои : 10.1021/ja9716721 .
- ^ Кримминс МТ; Чаудхари К. (2000). «Титановые еноляты хиральных вспомогательных веществ тиазолидинетиона: универсальные инструменты для асимметричного альдольного присоединения». Органические письма . 2 (6): 775–777. дои : 10.1021/ol9913901 . ПМИД 10754681 .
- ^ Кримминс, Майкл Т.; Шамзад, Мариам (2007). «Высокоселективные добавки ацетат-альдолов с использованием мезитилзамещенных хиральных вспомогательных веществ». Орг. Летт . 9 (1): 149–152. дои : 10.1021/ol062688b . ПМИД 17192107 .
- ^ СБ Дженнифер Кан; Кеннет К.-Х. Нг; Ян Патерсон (2013). «Влияние реакции Мукаяма-альдоль на полный синтез». Angewandte Chemie, международное издание . 52 (35): 9097–9108. дои : 10.1002/anie.201303914 . ПМИД 23893491 .
- ^ Теруаки Мукаяма; Кадзуо Банно; Коичи Нарасака (1974). «Реакции эфиров силилинолов с карбонильными соединениями, активированными тетрахлоридом титана». Журнал Американского химического общества . 96 (24): 7503–7509. дои : 10.1021/ja00831a019 .
- ^ 3-Гидрокси-3-метил-1-фенил-1-бутанон в результате перекрестной альдольной реакции Теруаки Мукаямы и Коичи Нарасаки Органический синтез , Coll. Том. 8, с.323 ( 1993 ); Том. 65, стр.6 ( 1987 )
- ^ Каррейра, ЕМ; Феттс, А.; Мартл, К. (2006). Каталитические энантиоселективные реакции альдольного присоединения . Орг. Реагировать. Том. 67. стр. 1–216. дои : 10.1002/0471264180.или067.01 . ISBN 978-0471264187 .
- ^ З.Г. Хайос, Д.Р. Пэрриш, патент Германии DE 2102623, 1971 г.
- ^ Хаджос, Золтан Г.; Пэрриш, Дэвид Р. (1974). «Асимметричный синтез бициклических промежуточных продуктов химии природных продуктов». Журнал органической химии . 39 (12): 1615–1621. дои : 10.1021/jo00925a003 .
- ^ Эдер, Ульрих; Зауэр, Герхард; Вихерт, Рудольф (1971). «Новый тип асимметричной циклизации оптически активных стероидных частичных структур CD». Angewandte Chemie International Edition на английском языке . 10 (7): 1615–1621. дои : 10.1002/anie.197104961 .
- ^ Нортруп, Алан Б.; Макмиллан Дэвид У.К. (2002). «Первая прямая и энантиоселективная перекрестно-альдольная реакция альдегидов» (PDF) . Журнал Американского химического общества . 124 (24): 6798–6799. дои : 10.1021/ja0262378 . ПМИД 12059180 .
- ^ Эванс, Пенсильвания ; Тедроу, Дж. С.; Шоу, Джей Ти; Дауни, CW (2002). «Диастереоселективные антиальдольные реакции хиральных N-ацилоксазолидинонов, катализируемые галогенидом магния». Журнал Американского химического общества . 124 (3): 392–393. дои : 10.1021/ja0119548 . ПМИД 11792206 .
- ^ Эванс, Дэвид А .; Дауни, К. Уэйд; Шоу, Джаред Т.; Тедроу, Джейсон С. (2002). «Катализируемые галогенидом магния антиальдольные реакции хиральных N-ацилтиазолидинтионов». Органические письма . 4 (7): 1127–1130. дои : 10.1021/ol025553o . ПМИД 11922799 .
- ^ Нортруп АБ; Мангион ИК; Хеттче Ф.; Макмиллан DWC (2004). «Энантиоселективные органокаталитические прямые альдольные реакции -оксиальдегидов: первый этап двухстадийного синтеза углеводов» . Angewandte Chemie International Edition на английском языке . 43 (16): 2152–2154. дои : 10.1002/anie.200453716 . ПМИД 15083470 .
- ↑ Бородин сообщил о конденсации пентаналя ( валерианальдегида ) с гептаналем ( энантальдегидом ) в: фон Рихтер, В. (1869) «В. фон Рихтер, из Петербурга 17 октября 1869 года» (В. фон Рихтер [репортаж] из Петербурга 17 октября 1869 г.), Отчеты Немецкого химического общества (на немецком языке), 2 : 552-553.
- Английская версия отчета Рихтера: (Сотрудники) (10 декабря 1869 г.) «Химические уведомления из иностранных источников: Отчеты Немецкого химического общества в Берлине, № 16, 1869 г.: Валериановый альдегид и энантальдегид - М. Бородин», The Chemical Новости и журнал промышленной науки , 20 : 286.
- ^ Гарнер, Сьюзан Эми (2007) «Образование углерод-углеродных связей при помощи водорода: применяется к восстановительным альдольным реакциям и реакциям Манниха», доктор философии. диссертация, Техасский университет (Остин), стр. 4 и 51.
- ^ Бородин, А. (1873) «О новом производном альдегида валерианы», отчеты Немецкого химического общества (на немецком языке), 6 : 982–985.
Дальнейшее чтение
[ редактировать ]- Chem 206, 215 Конспект лекций (2003, 2006) , Д.А. Эванса А.Г. Майерса и др. , Гарвардский университет (стр. 345, 936).