1,3-Диполярное циклоприсоединение
| 1,3-диполярное циклоприсоединение Хьюсгена | |
|---|---|
| Назван в честь | Рольф Хейсген |
| Тип реакции | Реакция образования кольца |
| Идентификаторы | |
| Портал органической химии | Хьюсген-1,3-диполярное циклоприсоединение |
| RSC Идентификатор онтологии | RXNO: 0000018 |
1,3 -диполярное циклоприсоединение представляет собой химическую реакцию между 1,3-диполем и диполярофилом с образованием пятичленного кольца. Самые ранние 1,3-диполярные циклоприсоединения были описаны в конце 19 - начале 20 века, после открытия 1,3-диполей. Механистические исследования и синтетические применения были созданы в 1960-х годах, в первую очередь благодаря работам Рольфа Хейсгена . [ 1 ] [ 2 ] Следовательно, реакцию иногда называют циклоприсоединением Хейсгена (этот термин часто используется для описания 1,3-диполярного циклоприсоединения между органическим азидом и алкином с образованием 1,2,3-триазола ). 1,3-диполярное циклоприсоединение — важный путь регио- и стереоселективного синтеза пятичленных гетероциклов и их ациклических производных с раскрытым кольцом. Диполярофил обычно представляет собой алкен или алкин, но может представлять собой и другие пи-системы. Когда диполярофил представляет собой алкин, обычно образуются ароматические кольца.

Обзор механики
[ редактировать ]Первоначально два предложенных механизма описывают 1,3-диполярное циклоприсоединение: во-первых, механизм согласованного перициклического циклоприсоединения , предложенный Рольфом Хейсгеном; [ 3 ] и, во-вторых, ступенчатый механизм с участием дирадикального промежуточного соединения , предложенный Файерстоуном. [ 4 ] После долгих дебатов первое предложение теперь общепринято. [ 5 ] - 1,3-диполь реагирует с диполярофилом согласованным , часто асинхронным и симметрии разрешенным по π 4 s + π 2 s образом через термическое шестиэлектронное ароматическое переходное состояние Хюккеля . Однако существует несколько примеров ступенчатого механизма реакций 1,3-диполярного циклоприсоединения тиокарбонилилидов без катализатора. [ 6 ] и оксиды нитрила [ 7 ]

Перициклический механизм
[ редактировать ]Хейсген исследовал серию циклоприсоединения между 1,3-диполярными диазосоединениями и различными диполярофильными алкенами . [ 3 ] Следующие наблюдения подтверждают согласованный перициклический механизм и опровергают ступенчатый дирадикальный или ступенчатый полярный путь.
- заместителей Эффекты : Различные заместители в диполе не оказывают большого влияния на скорость циклоприсоединения, что позволяет предположить, что в реакции не участвуют промежуточные соединения с разделенными зарядами.
- Эффекты растворителя : полярность растворителя мало влияет на скорость циклоприсоединения, что соответствует перициклическому механизму, при котором полярность не сильно меняется при переходе от реагентов к переходному состоянию.
- Стереохимия : 1,3-диполярные циклоприсоединения всегда стереоспецифичны по отношению к диполярофилу (т.е. цис -алкены, дающие син -продукты), поддерживая согласованный перициклический механизм, при котором две сигма-связи образуются одновременно.
- Термодинамические параметры : 1,3-диполярные циклоприсоединения имеют необычно большую отрицательную энтропию активации, аналогичную энтропии активации реакции Дильса-Альдера , что позволяет предположить, что переходное состояние высокоупорядочено, что является признаком согласованных перициклических реакций.
1,3-диполь
[ редактировать ]
1,3-диполь представляет собой органическую молекулу, которая может быть представлена в виде аллильного типа или пропаргильного / алленильного типа цвиттер-ионных октетных/секстетных структур . Оба типа 1,3-диполей имеют четыре электрона в π-системе на трех атомах. Аллиловый тип изогнут, тогда как пропаргил/алленильный тип имеет линейную геометрию . [ 8 ] 1,3-диполи, содержащие элементы более высокого ряда, такие как сера или фосфор, также известны, но используются реже.
Резонансные структуры могут быть нарисованы для делокализации как отрицательных, так и положительных зарядов на любом конце 1,3-диполя (см. схему ниже). Более точный метод описания электронного распределения в 1,3-диполе состоит в определении основного источника резонанса на основе экспериментальных или теоретических данных, таких как дипольного момента . измерения [ 9 ] или вычисления. [ 10 ] Например, диазометан несет наибольший отрицательный характер на концевом атоме азота, а азотистоводородная кислота имеет наибольший отрицательный характер на внутреннем атоме азота.

Следовательно, эта амбивалентность означает, что концы 1,3-диполя можно рассматривать как нуклеофильные и электрофильные одновременно. Степень нуклеофильности и электрофильности на каждом конце можно оценить с помощью граничных молекулярных орбиталей , которые можно получить вычислительным путем. В общем, атом, имеющий наибольший орбитальный коэффициент в ВЗМО, действует как нуклеофил, тогда как в НСМО действует как электрофил. Наиболее нуклеофильный атом обычно, но не всегда, является наиболее богатым электронами атомом. [ 11 ] [ 12 ] [ 13 ] В 1,3-диполярных циклоприсоединениях идентичность пары диполь-диполярофил определяет, будет ли доминировать ВЗМО или НСМО-характер 1,3-диполя (см. обсуждение пограничных молекулярных орбиталей ниже).
Диполярофил
[ редактировать ]Наиболее часто используемые диполярофилы — алкены и алкины. Диполярофилы, содержащие гетероатомы , такие как карбонилы и имины, также могут подвергаться 1,3-диполярному циклоприсоединению. Другие примеры диполярофилов включают фуллерены и нанотрубки , которые могут подвергаться 1,3-диполярному циклоприсоединению с азометинилидом в реакции Прато .
Эффекты растворителя
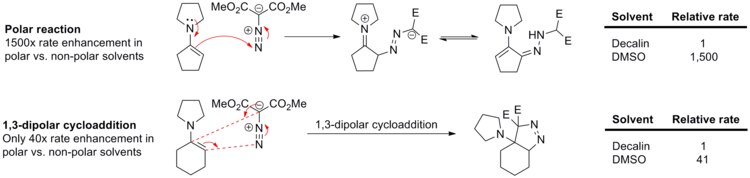
[ редактировать ]1,3-диполярные циклоприсоединения оказывают очень незначительное влияние растворителя, поскольку как реагенты, так и переходные состояния обычно неполярны. Например, скорость реакции фенилдиазометана с этилакрилатом или норборненом (см. схему ниже) меняется незначительно при изменении растворителя от циклогексана до метанола. [ 14 ]

Отсутствие эффектов растворителя при 1,3-диполярном циклоприсоединении наглядно демонстрируется в реакции енаминов с диметилдиазомалонатом (см. схему ниже). [ 15 ] реакция нуклеофильного присоединения N-циклопентенилпирролидина диазосоединениям к Полярная протекает в 1500 раз быстрее в полярном ДМСО , чем в неполярном декалине . С другой стороны, близкий аналог этой реакции, 1,3-диполярное циклоприсоединение N-циклогексенилпирролидина к диметилдиазомалонату, ускоряется в ДМСО лишь в 41 раз по сравнению с декалином.
Пограничная теория молекулярных орбиталей
[ редактировать ]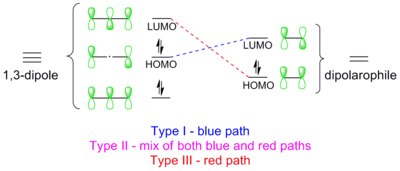
1,3-Диполярное циклоприсоединение представляет собой перициклическую реакцию, подчиняющуюся правилам Дьюара-Циммермана и правилам Вудворда-Гоффмана . В обработке Дьюара-Циммермана реакция протекает через 5-центровое, безузловое, 6-электронное переходное состояние Хюкеля для этой конкретной молекулярной орбитальной диаграммы. Однако каждой орбитали можно случайным образом присвоить знак, чтобы получить один и тот же результат. В трактовке Вудворда-Хоффмана граничные молекулярные орбитали (FMO) 1,3-диполя и диполярофила перекрываются в разрешенном симметрией порядке π 4 s + π 2 s . Такое перекрытие орбит может быть достигнуто тремя способами: типа I, II и III. [ 16 ] Доминирующим путем является тот, который имеет наименьшую энергетическую щель HOMO-LUMO.
Тип I
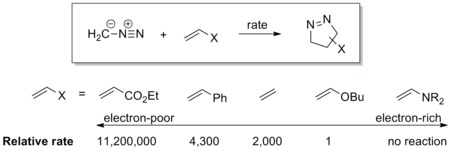
[ редактировать ]Диполь имеет высоколежащую ВЗМО , которая перекрывается с НСМО диполярофила. Диполь этого класса называется ВЗМО-управляемым диполем или нуклеофильным диполем , который включает азометин-илид , карбонилилид , нитрилилид , азометимин , карбонилимин и диазоалкан . Эти диполи легко присоединяются к электрофильным алкенам. Электроноакцепторные группы (EWG) на диполярофиле ускорят реакцию, снижая LUMO, тогда как электронодонорные группы (EDG) замедлят реакцию, повышая HOMO. Например, шкала реакционной способности диазометана по отношению к ряду диполярофилов представлена на схеме ниже. Диазометан реагирует с бедным электронами этилакрилатом более чем в миллион раз быстрее, чем богатый электронами бутилвиниловый эфир. [ 17 ]
Этот тип напоминает реакцию Дильса-Альдера с нормальной потребностью в электронах, в которой диеновая ВЗМО соединяется с диенофильной НСМО.
Тип II
[ редактировать ]ВЗМО диполя может образовывать пару с НСМО диполярофила; альтернативно, ВЗМО диполярофила может соединяться с НСМО диполя. Это двустороннее взаимодействие возникает потому, что энергетическая щель в обоих направлениях одинакова. Диполь этого класса называется HOMO-LUMO-контролируемым диполем или амбифильным диполем , который включает нитрилимид , нитрон , карбонилоксид , нитрилоксид и азид . Любой заместитель диполярофила ускорит реакцию за счет уменьшения энергетической щели между двумя взаимодействующими орбиталями; т. е. EWG снизит LUMO, а EDG повысит HOMO. Например, азиды реагируют с различными богатыми электронами и бедными электронами диполярофилами с одинаковой реакционной способностью (см. шкалу реакционной способности ниже). [ 18 ]

Тип III
[ редактировать ]Диполь имеет низколежащую НСМО, которая перекрывается с ВЗМО диполярофила (обозначена красными пунктирными линиями на диаграмме). Диполь этого класса называется LUMO-управляемым диполем или электрофильным диполем , в состав которого входят закись азота и озон . EWG на диполярофиле замедляют реакцию, а EDG ускоряют реакцию. Например, озон реагирует с богатым электронами 2-метилпропеном примерно в 100 000 раз быстрее, чем с бедным электронами тетрахлорэтеном (см. шкалу реакционной способности ниже). [ 19 ]
Этот тип напоминает реакцию Дильса-Альдера с обратной потребностью в электронах , в которой диеновая НСМО соединяется с диенофильной ВЗМО.

Реактивность
[ редактировать ]Согласованные процессы, такие как 1,3-циклоприсоединение, требуют высокоупорядоченного переходного состояния (высокая отрицательная энтропия активации) и лишь умеренных требований к энтальпии. С помощью экспериментов по конкурентным реакциям было обнаружено, что относительные скорости добавления для различных реакций циклоприсоединения позволяют сделать общие выводы о факторах реакционной способности.
- Сопряжение , особенно с ароматическими группами, увеличивает скорость реакции за счет стабилизации переходного состояния. Во время перехода две сигма-связи образуются с разной скоростью, что может генерировать частичные заряды в переходном состоянии, которые можно стабилизировать путем распределения заряда на сопряженные заместители.
- Более поляризуемые диполярофилы более реакционноспособны, поскольку диффузные электронные облака лучше подходят для инициирования потока электронов.
- Диполярофилы с высокой угловой деформацией более реакционноспособны из-за повышенной энергии основного состояния.
- Увеличение стерических затруднений в переходном состоянии в результате беспрепятственного взаимодействия реагентов резко снижает скорость реакции.
- Гетеродиполярофилы прибавляются медленнее, если вообще добавляются, по сравнению с C,C-диаполярофилами из-за меньшего прироста энергии сигма-связи, чтобы компенсировать потерю пи-связи во время переходного состояния.
- Изомерия диполярофила влияет на скорость реакции из-за стерики. транс -изомеры более реакционноспособны ( транс -стильбен присоединяется к дифенил(нитрилимиду) в 27 раз быстрее, чем цис -стильбен), потому что во время реакции валентный угол 120° сжимается до 109°, сближая затмевающие цис -заместители друг к другу для увеличения стерическое столкновение.

See Huisgen reference doi:10.1002/anie.196306331.

Стереоспецифичность
[ редактировать ]1,3-диполярное циклоприсоединение обычно приводит к сохранению конфигурации как по отношению к 1,3-диполю, так и по отношению к диполярофилу. Такая высокая степень стереоспецифичности является сильным подтверждением согласованности ступенчатых механизмов реакции. Как упоминалось ранее, многие примеры показывают, что реакции были ступенчатыми, таким образом, обладая частичной стереоспецифичностью или отсутствуя вообще.
По отношению к диполярофилу
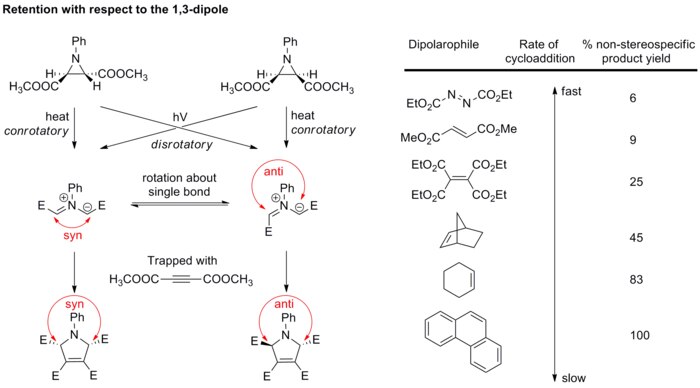
[ редактировать ]Цис -заместители в диполярофильном алкене превращаются в цис- , а транс -заместители в образующемся пятичленном циклическом соединении (см. схему ниже). [ 20 ]

По отношению к диполю
[ редактировать ]Как правило, стереохимия диполя не вызывает серьезного беспокойства, поскольку лишь немногие диполи могут образовывать стереогенные центры , а резонансные структуры допускают вращение связей, которое искажает стереохимию. Однако исследование азометинилидов подтвердило, что циклоприсоединение также стереоспецифично по отношению к дипольному компоненту. Диастереочистые азометинилиды образуются в результате электроциклического раскрытия кольца азиридинов , а затем быстро захватываются сильными диполярофилами , прежде чем может произойти вращение связей (см. схему ниже). [ 21 ] [ 22 ] Если используются более слабые диполярофилы, связи в диполе могут вращаться, что приводит к нарушению стереоспецифичности циклоприсоединения.
Эти результаты в целом подтверждают, что 1,3-диполярное циклоприсоединение является стереоспецифичным, обеспечивая сохранение как 1,3-диполя, так и диполярофила.
Диастереоселективность
[ редактировать ]Когда два или более стереоцентров в ходе реакции образуются , могут быть получены диастереомерные переходные состояния и продукты. При циклоприсоединении Дильса-Альдера эндодиастереоселективность , обусловленная вторичными орбитальными взаимодействиями обычно наблюдается . Однако в 1,3-диполярном циклоприсоединении на диастереоселективность влияют две силы: притягивающее π-взаимодействие (напоминающее вторичные орбитальные взаимодействия в циклоприсоединении Дильса-Альдера) и отталкивающее стерическое взаимодействие. К сожалению, эти две силы часто компенсируют друг друга, вызывая плохую диастереоселекцию при 1,3-диполярном циклоприсоединении.
Примеры субстрат-контролируемых диастереоселективных 1,3-диполярных циклоприсоединений показаны ниже. Сначала происходит реакция между бензонитрил-N-бензилидом и метилакрилатом . В переходном состоянии фенильные и метиловые эфирные группы складываются, образуя цис -замещение в качестве исключительного конечного пирролинового продукта. Это благоприятное π-взаимодействие компенсирует стерическое отталкивание между фенильными и метилэфирными группами. [ 23 ] Во-вторых, это реакция между нитроном и дигидрофураном . Экзо - селективность достигается для минимизации стерического отталкивания. [ 24 ] Последним является внутримолекулярная реакция азометинилида с алкеном. Диастереоселективность контролируется образованием менее напряженной цис - слитой кольцевой системы . [ 25 ]

Направленное 1,3-диполярное циклоприсоединение
[ редактировать ]Траекторией циклоприсоединения можно управлять для достижения диастереоселективной реакции. Например, металлы могут хелатировать диполярофил и входящий диполь и избирательно направлять циклоприсоединение на одну сторону. В приведенном ниже примере показано присоединение оксида нитрила к энантиомерно чистому аллиловому спирту в присутствии иона магния. В наиболее стабильной конформации алкена гидроксильная группа находится над плоскостью алкена. Затем магний хелатируется с гидроксильной группой и атомом кислорода оксида нитрила. Таким образом, циклоприсоединение избирательно происходит с верхней грани. [ 26 ]
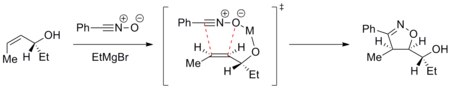
Такое диастереонаправление было применено при синтезе эпотилонов . [ 27 ]

Региоселективность
[ редактировать ]Для асимметричных пар диполь-диполярофил два региоизомерных продукта возможны . Как электронные/стереоэлектронные, так и стерические факторы способствуют региоселективности 1,3-диполярного циклоприсоединения. [ 28 ]
Электронный/стереоэлектронный эффект
[ редактировать ]Доминирующим электронным взаимодействием является комбинация крупнейшей ВЗМО и крупнейшей НСМО. Следовательно, региоселективность определяется атомами, имеющими наибольшие орбитальные коэффициенты ВЗМО и НСМО. [ 29 ] [ 30 ]
Например, рассмотрим циклоприсоединение диазометана к трем диполярофилам: метилакрилату , стиролу или метилциннамату . Углерод диазометана несет наибольшую ВЗМО, тогда как концевые олефиновые атомы углерода метилакрилата и стирола содержат наибольшую НСМО. Следовательно, циклоприсоединение дает региоселективное замещение в положении С-3. В случае метилциннамата два заместителя (Ph и COOMe) конкурируют за отрыв электронов от алкена. Карбоксил является лучшей электроноакцепторной группой, поэтому β-углерод является наиболее электрофильным. Таким образом, циклоприсоединение региоселективно дает карбоксильную группу на C-3 и фенильную группу на C-4.

Стерический эффект
[ редактировать ]Стерические эффекты могут как сотрудничать, так и конкурировать с вышеупомянутыми электронными эффектами. Иногда стерические эффекты полностью перевешивают электронное предпочтение, отдавая исключительно противоположный региоизомер. [ 31 ]
Например, диазометан обычно присоединяется к метилакрилату с образованием 3- карбоксилпиразолина . Однако, предъявляя к системе более стерические требования, мы начинаем наблюдать изомерные 4-карбоксильные пиразолины. Соотношение этих двух региоизомеров зависит от стерических требований. В крайнем случае, увеличение размера от водорода до трет-бутила сдвигает региоселективность со 100% 3-карбоксильного замещения на 100% 4-карбоксильного. [ 32 ] [ 33 ]

Синтетические приложения
[ редактировать ]1,3-диполярные циклоприсоединения являются важными путями синтеза многих важных 5-членных гетероциклов, таких как триазолы , фураны , изоксазолы , пирролидины и другие. Кроме того, некоторые циклоаддукты могут быть расщеплены, обнажая линейный скелет, открывая еще один путь к синтезу алифатических соединений . Эти реакции чрезвычайно полезны еще и потому, что они стереоспецифичны, диастереоселективны и региоселективны. Ниже приведены несколько примеров.
Оксиды нитрила
[ редактировать ]является 1,3-диполярное циклоприсоединение с нитрилоксидами Широко используемой маскированной альдольной реакцией . Циклоприсоединение между оксидом нитрила и алкеном дает циклический изоксазолин, тогда как реакция с алкином дает изоксазол. И изоксазолины, и изоксазолы могут быть расщеплены гидрированием с образованием β-гидроксикарбонильных продуктов альдольного типа или β-дикарбонильных продуктов кляйзеновского типа соответственно.
Циклоприсоединение нитрилоксид-алкин с последующим гидрированием было использовано в синтезе мияколида, как показано на рисунке ниже. [ 34 ]

Карбонилилиды
[ редактировать ]Реакции 1,3-диполярного циклоприсоединения стали мощным инструментом синтеза сложных циклических каркасов и молекул для медицинских, биологических и механистических исследований. Среди них реакции [3+2] циклоприсоединения с участием карбонилилидов широко используются для создания кислородсодержащих пятичленных циклических молекул. [ 35 ]
Получение карбонилилидов для реакций 1,3-диполярного циклоприсоединения.
[ редактировать ]Илиды рассматриваются как положительно заряженные гетероатомы , связанные с отрицательно заряженными атомами углерода, к которым относятся илиды сульфония , тиокарбонила , оксония , азота и карбонила . [ 36 ] Существует несколько методов получения карбонилилидов, которые являются необходимыми промежуточными продуктами для образования кислородсодержащих пятичленных кольцевых структур, для реакций [3+2] циклоприсоединения.
Синтез карбонилилидов из производных диазометана фотокатализом
[ редактировать ]карбонилилида Один из самых ранних примеров синтеза включает фотокатализ . [ 37 ] Фотолиз диазотетракис(трифторметил)циклопентадиена* (ДТТК) в присутствии тетраметилмочевины может привести к образованию карбонилилида путем межмолекулярной нуклеофильной атаки и последующей ароматизации фрагмента ДТТК. [ 37 ] Он был выделен и охарактеризован с помощью рентгеновской кристаллографии из-за стабильности, придаваемой ароматичностью, электроноакцепторными трифторметильными группами и электронодонорными диметиламиновыми группами. карбонилилида Стабильные диполи затем можно использовать в реакциях [3+2] циклоприсоединения с диполярофилами.

О другом раннем примере синтеза карбонилилида фотокатализом сообщили Olah et al . [ 38 ] Дидейтериодиазометан фотолизовали в присутствии формальдегида с образованием карбонилилида дидейтериоформальдегида.

Синтез карбонилилидов из гидроксипиронов методом переноса протона
[ редактировать ]Карбонилилиды могут быть синтезированы кислотным катализом гидрокси-3-пиронов в отсутствие металлического катализатора . [ 39 ] Происходит первоначальная таутомеризация , за которой следует удаление уходящей группы с ароматизацией пиронового кольца и образованием карбонилилида. В результате реакции циклоприсоединения с диполярофилом образуется оксацикл. Этот подход используется менее широко из-за его ограниченной применимости и потребности в пироновых скелетах.

5-гидрокси-4-пироны также можно использовать для синтеза карбонилилидов путем внутримолекулярного переноса водорода . [ 40 ] После переноса водорода карбонилилид может затем реагировать с диполярофилами с образованием кислородсодержащих колец.

Синтез α-галогенкарбонилилидов из дигалокарбенов
[ редактировать ]Дигалокарбены также использовались для получения карбонилилидов, используя электроноакцепторную природу дигалокарбенов. [ 41 ] [ 42 ] [ 43 ] И фенил(трихлорметил)ртуть, и фенил(трибромметил)ртуть являются источниками дихлоркарбенов и дибромкарбенов соответственно. Карбонилилид может быть получен при реакции дигалогенкарбенов с кетонами или альдегидами . Однако синтез α-галогенкарбонилилидов может также нежелательно приводить к потере монооксида углерода и образованию продукта дезоксигенирования.

Синтез карбонилилидов из производных диазометана металлокатализом
[ редактировать ]Универсальный подход к получению карбонилилидов включает металлический катализ α-диазокарбонильных соединений, обычно в присутствии димедных или диродиевых катализаторов. [ 44 ] После выделения газообразного азота и превращения в металлокарбен межмолекулярная реакция с карбонильной группой может привести к образованию карбонилилида. Последующая реакция циклоприсоединения с алкеном или алкиндиполярофилом может привести к образованию кислородсодержащих пятичленных колец. Популярные катализаторы, которые дают умеренный выход при синтезе оксациклов, включают Rh 2 (OAc) 4 и Cu(acac) 2 . [ 45 ] [ 46 ]

Механизм реакции 1,3-диполярного циклоприсоединения, опосредованной металлокализом диазокарбонильных соединений
[ редактировать ]Универсальность и широкое использование реакций 1,3-диполярного циклоприсоединения, опосредованных металлическим катализом молекул диазокарбонила, для синтеза кислородсодержащих пятичленных колец вызвали значительный интерес к ее механизму. Несколько групп исследовали механизм расширения возможностей синтетических молекул в отношении регио- и стереоселективности . Однако из-за высокой частоты повторения этих реакций промежуточные продукты и механизм остаются неуловимыми. Общепринятый механизм, разработанный при характеристике стабильных рутениево-карбеноидных комплексов. [ 47 ] и металлокарбены родия, [ 48 ] включает первоначальное образование комплекса металл-карбеноид из диазосоединения . Удаление газообразного азота приводит к образованию металлокарбена. Внутримолекулярная нуклеофильная атака карбонильного кислорода регенерирует металлический катализатор и образует карбонилилид. Затем карбонилилид может реагировать с алкеном или алкином, таким как диметилацетилендикарбоксилат (ДМАД), с образованием оксацикла.

Однако неясно, образует ли промежуточный металлокарбен карбонилилид. В некоторых случаях металлокарбены могут реагировать и непосредственно с диполярофилами. [ 49 ] В этих случаях металлокарбен, такой как карбен тетракарбоксилата диродия (II), стабилизируется посредством гиперконъюгативных металла взаимодействий типа енолята . [ 50 ] [ 51 ] [ 52 ] [ 53 ] Последующая реакция 1,3-диполярного циклоприсоединения происходит через переходный карбонилилид, образующий комплекс с металлом. Следовательно, стойкий металлокарбен может влиять на стереоселективность и региоселективность реакции 1,3-диполярного циклоприсоединения, основанную на стереохимии и размере металлических лигандов .

Механизм реакции 1,3-диполярного циклоприсоединения между диполем карбонилилида и алкинил- или алкенилдиполярофилами был тщательно исследован в отношении региоселективности и стереоселективности. Поскольку симметричные диполярофилы имеют одну ориентацию циклоприсоединения, можно получить только один региоизомер несколько стереоизомеров . , но можно получить [ 53 ] Напротив, несимметричные диполярофилы могут иметь множество региоизомеров и стереоизомеров. Эти региоизомеры и стереоизомеры могут быть предсказаны на основе теории пограничных молекулярных орбиталей (FMO) , стерических взаимодействий и стереоэлектронных взаимодействий . [ 54 ] [ 55 ]

Региоселективность реакции 1,3-диполярного циклоприсоединения, опосредованной металлокализом диазокарбонильных соединений
[ редактировать ]Региоселективность реакций 1,3-диполярного циклоприсоединения между диполями карбонилилида и алкинил- или алкенилдиполярофилами необходима для создания молекул с определенной региохимией. Теория FMO и анализ энергетических зазоров HOMO-LUMO между диполем и диполярофилом могут рационализировать и предсказать региоселективность экспериментальных результатов. [ 56 ] [ 57 ] ВЗМО и НСМО могут принадлежать либо к диполю, либо к диполярофилу, для которых ВЗМО -диполь - НСМО- диполярофил или ВЗМО- диполярофил - НСМО- диполь могут существовать взаимодействия . Перекрытие орбиталей с наибольшими коэффициентами может в конечном итоге рационализировать и предсказать результаты.

Архетипическая региоселективность реакции 1,3-диполярного циклоприсоединения, опосредованной диполями карбонилилида, была исследована Падвой и его сотрудниками. [ 55 ] [ 58 ] С помощью катализатора Rh 2 (OAc) 4 в бензоле диазодион подвергся реакции 1,3-диполярного циклоприсоединения с метилпропиолатом и метилпропаргиловым эфиром . Реакция с метилпропиолатом дает два региоизомера, основной из которых образуется в результате взаимодействия диполя ВЗМО - диполярофила НСМО , который имеет наибольшие коэффициенты при углероде, проксимальном к карбонильной группе карбонилилида, и при концевом алкиновом углероде метилпропиолата. Реакция с метилпропаргиловым эфиром дает один региоизомер, образующийся в результате взаимодействия ВЗМО -диполярофил - дипольного НСМО, который имеет наибольшие коэффициенты на углероде, дистальном по отношению к карбонильной группе карбонилилида, и на концевом алкиновом углероде метилпропаргилового эфира.
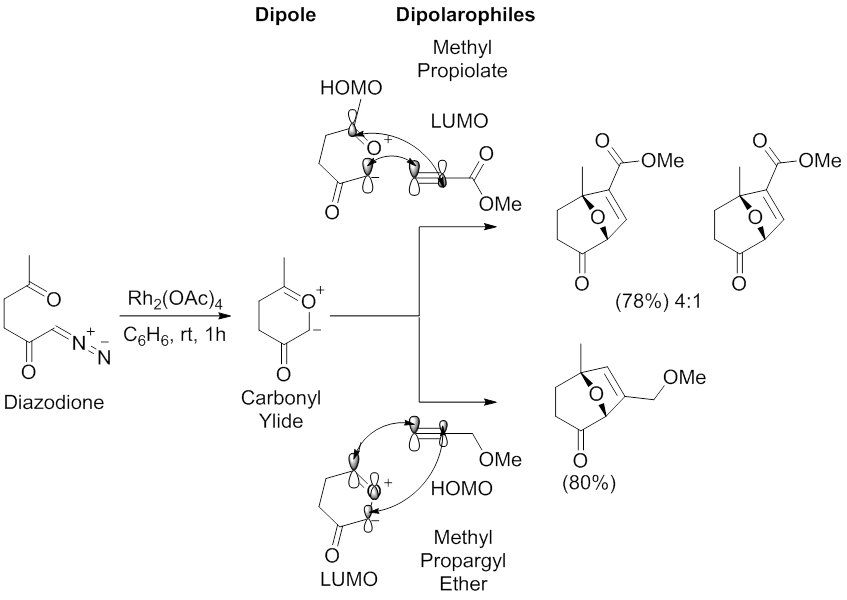
На региоселективность реакций 1,3-диполярного циклоприсоединения, опосредованных металлическим катализом диазокарбонильных соединений, также может влиять металл через образование стабильных металлокарбенов. [ 49 ] [ 59 ] Стабилизация металлокарбена посредством взаимодействий типа енолята металла предотвратит образование карбонилилидов, что приведет к прямой реакции между диполем металлокарбена и алкинил- или алкенилдиполярофилом (см. изображение металлокарбена тетракарбоксилата диродия (II), стабилизированного π C -Rh →π C=O гиперсопряжение.). В этой ситуации металлические лиганды будут влиять на региоселективность и стереоселективность реакции 1,3-диполярного циклоприсоединения.
Стереоселективность и асимметричная индукция реакции 1,3-диполярного циклоприсоединения, опосредованной металлокализом диазокарбонильных соединений
[ редактировать ]Стереоселективность . реакций 1,3-диполярного циклоприсоединения между диполями карбонилилида и алкенилдиполярофилами также была тщательно изучена Для алкинилдиполярофилов стереоселективность не является проблемой, поскольку относительно плоские sp 2 образуются углероды, при этом необходимо учитывать региоселективность (см. изображение продуктов реакции 1,3-диполярного циклоприсоединения между диполями карбонилилида и алкенил- или алкинилдиполярофилами). Однако для алкенилдиполярофилов как региоселективность, так и стереоселективность следует рассматривать как sp. 3 Углерод образуется в видах продукта.
Реакции 1,3-диполярного циклоприсоединения между диполями карбонилилида и алкенилдиполярофилами могут образовывать диастереомерные продукты. [ 53 ] Экзо - продукт характеризуется наличием диполярофильных заместителей, находящихся в цис-группе по отношению к эфирному мостику оксацикла. Эндо - продукт характеризуется тем, что диполярофильные заместители расположены транс по отношению к эфирному мостику оксацикла. Оба продукта могут быть созданы посредством перициклических переходных состояний, включающих согласованные синхронные или согласованные асинхронные процессы.
Один из ранних примеров обеспечивал стереоселективность в отношении эндо- и экзопродуктов с металлическими катализаторами и кислотами Льюиса. [ 60 ] Реакции только с металлическим катализатором Rh 2 (OAc) 4 отдают предпочтение экзо -продукту, тогда как реакции с дополнительной кислотой Льюиса Yb(OTf) 3 предпочитают эндо- продукт. Эндоселективность , наблюдаемая в реакциях циклоприсоединения кислот Льюиса, объясняется оптимизированным перекрытием орбиталей карбонильных π-систем между диполярофилом, координируемым Yb(Otf) 3 (LUMO), и диполем (HOMO). После многих исследований были разработаны два основных подхода к влиянию на стереоселективность циклоприсоединения карбонилилида, использующие хиральность металлических катализаторов и кислот Льюиса. [ 53 ]

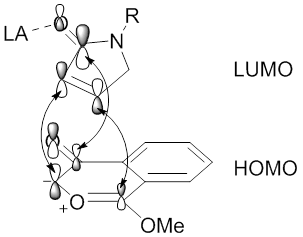
В первом подходе используются хиральные металлические катализаторы для модуляции эндо- и экзо -стереоселективности. Хиральные катализаторы, в частности Rh 2 [( S )-DOSP] 4 и Rh 2 [( S )-BPTV] 4, могут вызывать умеренную асимметричную индукцию и были использованы для синтеза противогрибкового агента псевдоларовой кислоты А. [ 61 ] Это результат того, что хиральный металлический катализатор остается связанным с карбонилилидом во время циклоприсоединения, что придает селективность по граням. Однако точные механизмы еще не до конца изучены.

Во втором подходе используется хиральный катализатор на основе кислоты Льюиса для индукции лицевой стереоселективности после образования карбонилилида с использованием ахирального металлического катализатора. [ 62 ] Считается, что хиральный катализатор на основе кислоты Льюиса координируется с диполярофилом, что снижает НСМО диполярофила, а также приводит к энантиоселективности .

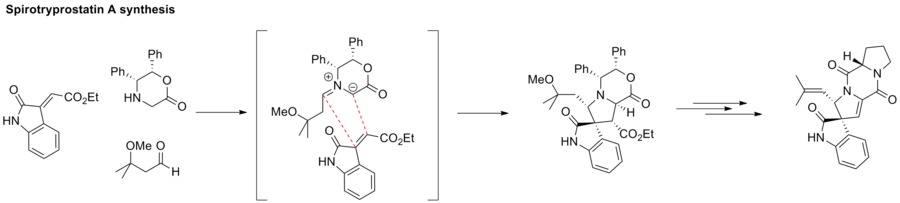
Азометинилиды
[ редактировать ]1,3-Диполярное циклоприсоединение между азометинилидом и алкеном образует азациклическую структуру, такую как пирролидин . Эта стратегия была применена к синтезу спиротрипростатина А. [ 63 ]
Озон
[ редактировать ]Озонолиз – очень важная органическая реакция. Алкены и алкины могут расщепляться озонолизом с образованием альдегидов , кетонов или карбоновых кислот .
Биологические применения
[ редактировать ]1,3-диполярное циклоприсоединение между органическими азидами и концевыми алкинами (т.е. циклоприсоединение Хейсгена ) широко используется для биоконъюгации .
Медный катализ
[ редактировать ]Реакция Хейсгена обычно не протекает легко в мягких условиях. Мелдал и др. и Шарплесс и др. независимо разработал катализируемую медью (I) версию реакции Хейсгена, CuAAC (для катализируемого медью азид-алкинового циклоприсоединения), которая очень легко протекает в мягких, в том числе физиологических , условиях (нейтральный pH , комнатная температура и водный раствор ). [ 64 ] [ 65 ] Эта реакция также является биоортогональной : азиды и алкины обычно отсутствуют в биологических системах, и поэтому эти функциональные группы могут подвергаться хемоселективной реакции даже в клеточном контексте . Они также не вступают в реакцию с другими функциональными группами, встречающимися в природе, поэтому не нарушают биологические системы. Реакция настолько разнообразна, что ее называют химией «Клик» . Хотя медь(I) токсична , было разработано множество защитных лигандов как для снижения цитотоксичности, так и для повышения уровня CuAAC, что позволяет использовать ее в исследованиях in vivo . [ 66 ]

Например, Бертоцци и др. сообщили о метаболическом включении азид-функционализированных сахаридов в гликан клеточной мембраны и последующем мечении конъюгатом флуорофор -алкин. В результате клеточная мембрана флуоресцентно помечена и, следовательно, может быть отображена с помощью флуоресцентного микроскопа . [ 67 ]

Циклоприсоединение, стимулируемое штаммом
[ редактировать ]Чтобы избежать токсичности меди(I), Bertozzi et al. разработали азид-алкиновое циклоприсоединение, стимулируемое штаммом (SPAAC), между органическим азидом и напряженным циклооктином . Угловое искажение циклооктина помогает ускорить реакцию как за счет снижения напряжения активации, так и за счет усиления взаимодействия, что позволяет использовать его в физиологических условиях без необходимости использования катализатора. [ 68 ]

Например, Тинг и др. ввели азидофункциональность в специфические белки на поверхности клетки с помощью фермента лигазы . Затем меченный азидом белок метят конъюгатом циклооктин-флуорофор с получением флуоресцентно меченного белка. [ 69 ]

Ссылки
[ редактировать ]- ^ Бертран, Гай; Вентрап, Курт (17 марта 1994 г.). «Нитрилимины: от характеристики матрицы к стабильным соединениям». Angewandte Chemie International Edition на английском языке . 33 (5): 527–545. дои : 10.1002/anie.199405271 .
- ^ Хейсген, Рольф (октябрь 1963 г.). «1,3-Диполярные циклоприсоединения. Прошлое и будущее». Angewandte Chemie International Edition на английском языке . 2 (10): 565–598. дои : 10.1002/anie.196305651 .
- ^ Jump up to: а б Хейсген, Рольф (ноябрь 1963 г.). «Кинетика и механизм 1,3-диполярного циклоприсоединения». Angewandte Chemie, международное издание . 2 (11): 633–645. дои : 10.1002/anie.196306331 .
- ^ Файерстоун, Р. (1968). «Механизм 1,3-диполярного циклоприсоединения». Журнал органической химии . 33 (6): 2285–2290. дои : 10.1021/jo01270a023 .
- ^ Хейсген, Рольф (1976). «1,3-Диполярное циклоприсоединение. 76. Согласованная природа 1,3-диполярного циклоприсоединения и вопрос о дирадикальных интермедиатах». Журнал органической химии . 41 (3): 403–419. дои : 10.1021/jo00865a001 .
- ^ Млостон, Г.; Лангалс, Э.; Хейсген, Рольф (1986). «Первые двухступенчатые 1,2-диполярные циклоаддитоны: нестереоспецифичность». Дж. Ам. хим. Соц. 108 (20): 6401–66402. дои : 10.1021/ja00280a053 .
- ^ Сейед Амир, Сиадати (2015). «Пример ступенчатого механизма безкатализаторного 1,3-диполярного циклоприсоединения между оксидом нитрила и богатым электронами алкеном». Буквы тетраэдра . 56 (34): 4857–4863. дои : 10.1016/j.tetlet.2015.06.048 .
- ^ Хейсген, Рольф (1963). «1,3-Диполярные циклоприсоединения. Прошлое и будущее». Angewandte Chemie, международное издание . 2 (10): 565–598. дои : 10.1002/anie.196305651 .
- ^ Кокс, А; Томас, Л; Шеридан, Дж (1958). «Микроволновые спектры диазометана и его дейтеропроизводных». Природа . 181 (4614): 1000–1001. Бибкод : 1958Natur.181.1000C . дои : 10.1038/1811000a0 . S2CID 4245746 .
- ^ Хилберти, П; Лефорестье, К. (1978). «Разложение волновых функций молекулярных орбиталей в волновые функции валентных связей. Упрощенная процедура». Журнал Американского химического общества . 100 (7): 2012–2017. дои : 10.1021/ja00475a007 .
- ^ МакГаррити, Дж. Ф.; Патай, Саул (1978). «Основность, кислотность и водородная связь». Диазоний и диазогруппы . Том. 1. С. 179–230. дои : 10.1002/9780470771549.ch6 . ISBN 9780470771549 .
- ^ Бернер, Дэниел; МакГаррити, Джон (1979). «Прямое наблюдение иона метилдиазония во фторсерной кислоте». Журнал Американского химического общества . 101 (11): 3135–3136. дои : 10.1021/ja00505a059 .
- ^ Мюллер, Юджин; Рундель, Вольфганс (1956). «Исследования по диазометанам, VI. Сообщение: Реакция диазоэтана с метиллитием». Химические отчеты . 89 (4): 1065–1071. дои : 10.1002/cber.19560890436 .
- ^ Гайттнер, Йохен; Хейсген, Рольф; Рейссиг, Ганс-Ульрих (1978). «Зависимость скорости циклоприсоединения фенилдиазометана от растворителя и параметров активации» . Гетероциклы . 11 : 109–120. дои : 10.3987/S(N)-1978-01-0109 .
- ^ Хейсген, Рольф; Рейссиг, Ганс-Ульрих; Хубер, Гельмут; Восс, Сабина (1979). «α-Диазокарбонильные соединения и енамины - дихотомия путей реакции». Буквы тетраэдра . 20 (32): 2987–2990. дои : 10.1016/S0040-4039(00)70991-9 .
- ^ Сустманн, Р. (1974). «Орбитальный энергетический контроль реакционной способности циклоприсоединения» . Чистая и прикладная химия . 40 (4): 569–593. дои : 10.1351/pac197440040569 .
- ^ Гайттнер, Йохен; Хейсген, Рольф (1977). «Кинетика реакций 1,3-диполярного циклоприсоединения диазометана; корреляция с гомо-люминесцентными энергиями». Буквы тетраэдра . 18 (10): 881–884. дои : 10.1016/S0040-4039(01)92781-9 .
- ^ Хейсген, Рольф; Сеймис, Гюнтер; Мебиус, Леандер (1967). «K1.3-Диполярные циклоприсоединения, XXXII. Кинетика присоединения органических азидов к кратным связям CC». Химические отчеты . 100 (8): 2494–2507. дои : 10.1002/cber.19671000806 .
- ^ Уильямсон, генеральный директор; Цветанович, Р.Дж. (1968). «Скорости озон-олефиновых реакций в растворах четыреххлористого углерода». Журнал Американского химического общества . 90 (14): 3668–3672. дои : 10.1021/ja01016a011 .
- ^ Бильмайер, Вернер; Гайттнер, Йохен; Хейсген, Рольф; РейссигП, Ганс-Ульрих (1978). «Стереоспецифичность диазометановых циклоприсоединений» . Гетероциклы . 10 : 147–152. дои : 10.3987/S-1978-01-0147 .
- ^ Хейсген, Рольф; Шеер, Вольфганг; Хубер, Гельмут (1967). «Стереоспецифическое превращение цис-транс-изомерных азиридинов в азометин-илиды с открытой цепью». Журнал Американского химического общества . 89 (7): 1753–1755. дои : 10.1021/ja00983a052 .
- ^ Дамен, Александр; Хамбергер, Хельмут; Хейсген, Рольф; Марковски, Волкер (1971). «Открытие вращательного кольца оксидов цианостильбена до карбонилилидов». Журнал Химического общества D: Химические коммуникации (19): 1192–1194. дои : 10.1039/C29710001192 .
- ^ Падва, Альберт; Смоланофф, Джоэл (1971). «Фотоциклоприсоединение арилазиренов с электронодефицитными олефинами». Журнал Американского химического общества . 93 (2): 548–550. дои : 10.1021/ja00731a056 .
- ^ Какисава, Хироши (1979). Синтез dl-изоретронеканола» . Письма по химии . 8 11): 1337–1340 ( Ивашита, Такаши ; « .
- ^ Ван, Цзя-Линь; Рипка, Уильям; Конфалоне, Пэт (1984). «Краткий и стереоспецифический синтез (±)-α-ликорана». Буквы тетраэдра . 25 (41): 4613–4616. дои : 10.1016/S0040-4039(01)91213-4 .
- ^ Канемаса, Сюдзи (2002). «Стереоконтроль с помощью металлов реакций 1,3-диполярного циклоприсоединения». Синлетт . 2002 (9): 1371–1387. дои : 10.1055/s-2002-33506 .
- ^ Боде, Джеффри; Каррейра, Эрик (2011). «Стереоселективный синтез эпотилонов A и B посредством направленного циклоприсоединения оксида нитрила». Журнал Американского химического общества . 123 (15): 3611–3612. дои : 10.1021/ja0155635 . ПМИД 11472140 .
- ^ Всеволод Васильевич Ростовцев; Люк Г. Грин; Валерий В. Фокин; К. Барри Шарплесс (2002). «Пошаговый процесс циклоприсоединения Хейсгена: катализируемое медью (I) региоселективное лигирование азидов и терминальных алкинов». Angewandte Chemie, международное издание . 41 (14): 2596–22599. doi : 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4 . ПМИД 12203546 .
- ^ Карамелла, Пьерлуиджи; Хоук, КН (1976). «Геометрии бетаинов нитрилия. Выяснение явно аномальных реакций 1,3-диполей». Журнал Американского химического общества . 98 (20): 6397–6399. дои : 10.1021/ja00436a062 .
- ^ Карамель, Пьерлуиджи; Гандур, Рут В.; Холл, Джанет А.; Девиль, Синтия Г.; Хоук, К.Н. (1977). «Вывод формы и энергии молекулярных орбиталей 1,3-диполей. Оптимизация геометрии этих видов с помощью MINDO/2 и MINDO/3». Журнал Американского химического общества . 99 (2): 385–392. дои : 10.1021/ja00444a013 .
- ^ Хейсген, Рольф (ноябрь 1963 г.). «Кинетика и механизм 1,3-диполярного циклоприсоединения». Angewandte Chemie, международное издание . 2 (11): 633–645. дои : 10.1002/anie.196306331 .
- ^ Падва, Альберт (1983). Химия 1,3-диполярного циклоприсоединения . Серия «Общая гетероциклическая химия». Том. 1. Соединенные Штаты Америки: Wiley-Interscience. стр. 141–145. ISBN 978-0-471-08364-1 .
- ^ Кошиновский, Дж. (1980). диссертация (PhD Thesis).
- ^ Эванс, Дэвид; Рипин, Дэвид; Холстед, Дэвид; Кампос, Кевин (1999). «Синтез и абсолютное стереохимическое отнесение (+)-миаколида». Журнал Американского химического общества . 121 (29): 6816–6826. дои : 10.1021/ja990789h .
- ^ Синтетические реакции связей M = C и M = N: образование илида, перегруппировка и 1,3-диполярное циклоприсоединение; Хияма, Т.В., Дж., Ред.; Эльзевир, 2007; Том. 11.
- ^ Падва, Альберт; Хорнбакл, Сьюзен Ф. (1991). «Образование илида в результате реакции карбенов и карбеноидов с неподеленными парами гетероатомов». Химические обзоры . 91 (3): 263–309. дои : 10.1021/cr00003a001 .
- ^ Jump up to: а б Янулис, Евгений П.; Ардуэнго, Энтони Дж. (1983). «Структура электронно стабилизированного карбонилилида». Журнал Американского химического общества . 105 (18): 5929–5930. дои : 10.1021/ja00356a044 .
- ^ Пракаш, ГКС; Эллис, RW; Фельберг, доктор медицинских наук; Ола, Г.А. Формальдегид-0-метилид, [CH2=O+-CH2 : Исходный карбонилилид] J Am Chem Soc 1986, 108, 1341.
- ^ Сэммс, П.Г.; Стрит, Л.Дж. Внутримолекулярные циклоприсоединения с оксидопирилий-илидами J. Chem. Соц., хим. Коммун. 1982, 1056.
- ^ Гарст, Мэн; Макбрайд, Би Джей; Дуглас III, Дж. Г. Внутримолекулярные циклоприсоединения с 2-(ω-алкенил)-5-гидрокси-4-пиронами Tetrahedron Lett. 1983, 24, 1675.
- ^ Гиш, Джон Ф.; Ландгребе, Джон А. (1985). «Дихлоркарбен в результате флэш-вакуумного пиролиза триметил(трихлорметил)силана. Возможное наблюдение 1,1-дихлор-3-фенилкарбонилилида» . Журнал органической химии . 50 (12): 2050–2054. дои : 10.1021/jo00212a009 .
- ^ Хуан, Чжэньвэй; Ландгребе, Джон А.; Петерсон, Кимберли (1983). «Дибромкарбонилилиды. Деоксигенирование альдегидов и кетонов дибромкарбеном». Журнал органической химии . 48 (24): 4519–4523. дои : 10.1021/jo00172a015 .
- ^ Мартин, Чарльз В.; Лунд, Пол Р.; Рапп, Эрих; Ландгребе, Джон А. (1978). «Галогенированные карбонилилиды в реакциях предшественников ртутных дигалогенкарбенов с замещенными бензальдегидами». Журнал органической химии . 43 (6): 1071–1076. дои : 10.1021/jo00400a009 .
- ^ Ходжсон, DM; Брюкл, Т.; Глен, Р.; Лабанде, АХ; Селден, Д.А.; Доссеттер, АГ; Редгрейв, А.Дж. Каталитические энантиоселективные межмолекулярные циклоприсоединения карбонилилидов, производных 2-диазо-3,6-дикетоэфиров, с алкендиполярофилами, Труды Национальной академии наук Соединенных Штатов Америки, 2004, 101, 5450.
- ^ Падва, Альберт; Герцог, Дональд Л.; Надлер, Уильям Р. (1994). «Внутримолекулярное циклоприсоединение изомунконных диполей к гетероароматическим пи-системам». Журнал органической химии . 59 (23): 7072–7084. дои : 10.1021/jo00102a037 .
- ^ Хамагучи, М.; Ибата, Т. Новый тип мезойной системы. 1,3-Диполярное циклоприсоединение изомунчнона с этиленовыми соединениями Chem Lett 1975, 499.
- ^ Пак, Сун-Бонг; Саката, Наоя; Нисияма, Хисао (1996). «Арилоксикарбонилкарбеновые комплексы бис (оксазолинил) пиридинрутения как активные интермедиаты в асимметричных каталитических циклопропанациях». Химия - Европейский журнал . 2 (3): 303–306. дои : 10.1002/chem.19960020311 .
- ^ Снайдер, Джеймс П.; Падва, Альберт; Стенгель, Томас; Ардуенго, Энтони Дж.; Йокиш, Александр; Ким, Хё Чжун (2001). «Стабильный карбеноид тетракарбоксилата диродия: кристаллическая структура, анализ связей и катализ». Журнал Американского химического общества . 123 (45): 11318–11319. дои : 10.1021/ja016928o . ПМИД 11697986 .
- ^ Jump up to: а б Ходжсон, DM; Пьерар, FYTM; Стиппл, П.А. Каталитические энантиоселективные перегруппировки и циклоприсоединения с участием илидов диазосоединений Chem Soc Rev 2001, 30, 50.
- ^ Ёсикай, Наохико; Накамура, Эйичи (2003). «Теоретические исследования диастерео- и энантиоселективной родий-катализируемой циклизации диазосоединения посредством внутримолекулярного внедрения связи C—H». Расширенный синтез и катализ . 345 (910): 1159–1171. дои : 10.1002/adsc.200303092 .
- ^ Накамура, Эйичи; Ёсикай, Наохико; Яманака, Масахиро (2002). «Механизм активации связи C-H/реакция образования связи C-C между диазосоединением и алканом, катализируемая тетракарбоксилатом диродия». Журнал Американского химического общества . 124 (24): 7181–7192. дои : 10.1021/ja017823o . ПМИД 12059244 .
- ^ Константино, Г.; Ровито, Р.; Маккиаруло, А.; Пелличиари, Р. Структура металл-карбеноидных промежуточных продуктов, полученных в результате разложения α-диазокарбонильных соединений, опосредованного тетракарбоксилатом диродия (II): исследование DFT, J Mol Struc-Theochem 2002, 581, 111.
- ^ Jump up to: а б с д М. Ходжсон, Д.; Х. Лабанде, А.; Мутусами, С. В органических реакциях; Джон Вили и сыновья, Inc.: 2004.
- ^ Суга, Хироюки; Эбиура, Ясутака; Фукусима, Кадзуаки; Какехи, Акиказу; Баба, Тосихидэ (2005). «Эффективные каталитические эффекты кислот Льюиса в реакциях 1,3-диполярного циклоприсоединения карбонилилидов с иминами». Журнал органической химии . 70 (26): 10782–10791. дои : 10.1021/jo051743b . ПМИД 16356001 .
- ^ Jump up to: а б Падва, Альберт; Фрикселл, Глен Э.; Чжи, Линь (1990). «Тандемная реакция циклизации-циклоприсоединения карбеноидов родия. Область применения и механизмы процесса». Журнал Американского химического общества . 112 (8): 3100–3109. дои : 10.1021/ja00164a034 .
- ^ Хоук, КН; Симс, Джойнер; Герцог, RE; Строзье, RW; Джордж, Джон К. (1973). «Пограничные молекулярные орбитали 1,3-диполей и диполярофилов». Журнал Американского химического общества . 95 (22): 7287–7301. дои : 10.1021/ja00803a017 .
- ^ Хоук, КН; Рондан, Нельсон Г.; Сантьяго, Сьело; Галло, Кэтрин Дж.; Гандур, Рут Уэллс; Гриффин, Гэри В. (1980). «Теоретические исследования строения и реакций замещенных карбонилилидов». Журнал Американского химического общества . 102 (5): 1504–1512. дои : 10.1021/ja00525a006 .
- ^ Падва, Альберт; Вайнгартен, М. Дэвид (1996). «Каскадные процессы металлокарбеноидов». Химические обзоры . 96 (1): 223–270. дои : 10.1021/cr950022h . ПМИД 11848752 .
- ^ Падва, Альберт; Остин, Дэвид Дж. (1996). «Индуцированная лигандами селективность в реакциях α-диазокарбонильных соединений, катализируемых родием (II) †». Журнал органической химии . 61 : 63–72. дои : 10.1021/jo951576n .
- ^ Шуга, Х.; Какехи, А.; Ито, С.; Иноуэ, К.; Исида, Х.; Ибата, Т. Стереоконтроль в катализируемой трифлатом иттербия реакции 1,3-диполярного циклоприсоединения карбонилилида с N-замещенными малеимидами и диметилфумаратом B Chem Soc Jpn 2001, 74, 1115.
- ^ Гэн, Чжэ; Чен, Бин; Чиу, Полина (2006). «Полный синтез псевдоляровой кислоты А». Angewandte Chemie, международное издание . 45 (37): 6197–6201. дои : 10.1002/anie.200602056 . ПМИД 16906616 .
- ^ Суга, Хироюки; Иноуэ, Кей; Иноуэ, Шуичи; Какехи, Акиказу; Широ, Мотоо (2005). «Хиральные комплексы 2,6-бис(оксазолинил)пиридин-редкоземельных металлов как катализаторы высокоэнантиоселективных реакций 1,3-диполярного циклоприсоединения 2-бензопирилий-4-олатов». Журнал органической химии . 70 (1): 47–56. дои : 10.1021/jo049007f . ПМИД 15624905 .
- ^ Ониси, Томоюки; Себахар, Пол; Уильямс, Роберт (2003). «Краткий асимметричный полный синтез спиротрипростатина А». Органические письма . 5 (17): 3135–3137. дои : 10.1021/ol0351910 . ПМИД 12917000 .
- ^ Торное, Кристиан; Кристенсен, Каспар; Мелдал, Мортен (2002). «Пептидотриазолы на твердой фазе: [1,2,3]-триазолы путем региоспецифического катализируемого медью (I) 1,3-диполярного циклоприсоединения концевых алкинов к азидам». Журнал органической химии . 67 (9): 3057–3064. дои : 10.1021/jo011148j . ПМИД 11975567 . S2CID 11957672 .
- ^ Ростовцев, Всеволод; Грин, Люк; Фокин, Валерий; Шарплесс, Барри К. (2002). «Пошаговый процесс циклоприсоединения Хейсгена: катализируемое медью (I) региоселективное лигирование азидов и терминальных алкинов». Angewandte Chemie, международное издание . 41 (14): 2596–2599. doi : 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4 . ПМИД 12203546 .
- ^ Безансени-Веблер, Кристен; Цзян, Хао; Чжэн, Тяньцин; Фэн, Лей; Сориано дель Амо, Дэвид; Ван, Вэй; Кливанский, Лиана М.; Марлоу, Флоренс Л.; Лю, Йи; Ву, Пэн (2011). «Повышение эффективности биоортогональных реакций щелчка для биоконъюгации: сравнительное исследование» . Angewandte Chemie, международное издание . 50 (35): 8051–8056. дои : 10.1002/anie.201101817 . ПМЦ 3465470 . ПМИД 21761519 .
- ^ Брайденбах, Марк; Галлахер, Дженнифер; Король, Дэвид; Умный, Брайан; Ву, Пэн; Бертоцци, Кэролин (2010). «Целевая метаболическая маркировка дрожжевых N-гликанов неприродными сахарами» . Труды Национальной академии наук Соединенных Штатов Америки . 107 (9): 3988–3993. Бибкод : 2010PNAS..107.3988B . дои : 10.1073/pnas.0911247107 . ПМК 2840165 . ПМИД 20142501 .
- ^ Агард, Николас; Прешер, Дженнифер; Бертоцци, Кэролайн (2004). «Стимулируемое штаммом [3 + 2] азид-алкиновое циклоприсоединение для ковалентной модификации биомолекул в живых системах». Журнал Американского химического общества . 126 (46): 15046–15047. дои : 10.1021/ja044996f . ПМИД 15547999 .
- ^ Фернандес-Суарес, Марта; Баруа, Хеманта; Мартинес-Эрнандес, Лаура; Се, Кэтлин; Баскин, Джереми; Бертоцци, Кэролайн; Тинг, Алиса (2007). «Перенаправление лигазы липоевой кислоты для маркировки белков клеточной поверхности с помощью низкомолекулярных зондов» . Природная биотехнология . 25 (12): 1483–1487. дои : 10.1038/nbt1355 . ПМЦ 2654346 . ПМИД 18059260 .