Генетическое заболевание
| Генетическое заболевание | |
|---|---|
 | |
| Схема с примерами заболеваний, локализованных в каждой хромосоме. | |
| Специальность | Медицинская генетика |
Генетическое заболевание – это проблема со здоровьем, вызванная одной или несколькими аномалиями в геноме . Это может быть вызвано мутацией одного гена ( моногенный) или нескольких генов (полигенный) или хромосомной аномалией . Хотя полигенные заболевания являются наиболее распространенными, этот термин чаще всего используется при обсуждении заболеваний, имеющих единственную генетическую причину, либо в гене, либо в хромосоме . [1] [2] Ответственная мутация может возникнуть спонтанно до эмбрионального развития ( мутация de novo ) или может быть унаследована от двух родителей, являющихся носителями дефектного гена ( аутосомно-рецессивное наследование), или от родителя с заболеванием ( аутосомно-доминантное наследование). Когда генетическое заболевание унаследовано от одного или обоих родителей, оно также классифицируется как наследственное заболевание . Некоторые заболевания вызваны мутацией Х-хромосомы и имеют Х-сцепленное наследование. Очень немногие заболевания наследуются по Y-хромосоме или митохондриальной ДНК (из-за их размера). [3]
Известно более 6000 генетических нарушений. [4] в медицинской литературе постоянно описываются новые генетические нарушения. [5] Более 600 генетических нарушений поддаются лечению. [6] Примерно 1 из 50 человек страдает известным моногенным заболеванием, а около 1 из 263 страдает хромосомным заболеванием . [7] Около 65% людей имеют те или иные проблемы со здоровьем в результате врожденных генетических мутаций. [7] Из-за значительного количества генетических нарушений примерно 1 из 21 человека страдает генетическим заболеванием, классифицируемым как « редкое » (обычно затрагивающее менее 1 из 2000 человек). Большинство генетических нарушений сами по себе редки. [5] [8]
Генетические нарушения присутствуют еще до рождения, а некоторые генетические нарушения вызывают врожденные дефекты , но врожденные дефекты также могут быть скорее связанными с развитием, чем наследственными . Противоположностью наследственной болезни является приобретенная болезнь . Большинство видов рака , хотя они и связаны с генетическими мутациями небольшой части клеток организма, являются приобретенными заболеваниями. Однако некоторые раковые синдромы , такие как BRCA мутации , являются наследственными генетическими нарушениями. [9]
Одногенный
[ редактировать ]| Распространенность расстройства (приблизительно) | |
|---|---|
| Аутосомно-доминантный | |
| Семейная гиперхолестеринемия | 1 из 500 [11] |
| Миотоническая дистрофия 1 типа | 1 из 2100 [12] |
| Нейрофиброматоз I типа | 1 из 2500 [13] |
| Наследственный сфероцитоз | 1 из 5000 |
| синдром Марфана | 1 из 4000 [14] |
| болезнь Хантингтона | 1 из 15 000 [15] |
| Аутосомно-рецессивный | |
| Серповидноклеточная анемия | 1 из 625 [16] |
| Муковисцидоз | 1 из 2000 |
| Болезнь Тея-Сакса | 1 из 3000 |
| Фенилкетонурия | 1 из 12 000 |
| Аутосомно-рецессивный поликистоз почек | 1 из 20 000 [17] |
| Мукополисахаридозы | 1 из 25 000 |
| Дефицит лизосомальной кислой липазы | 1 из 40 000 |
| Болезни накопления гликогена | 1 из 50 000 |
| Галактоземия | 1 из 57 000 |
| X-связанный | |
| мышечная дистрофия Дюшенна | 1 из 5000 |
| Гемофилия | 1 из 10 000 |
| Значения указаны для живорожденных детей. | |
Одногенное заболевание (или моногенное заболевание ) является результатом мутации одного гена. Одногенные расстройства могут передаваться последующим поколениям несколькими способами. геномный импринтинг и однородительская дисомия Однако могут влиять на характер наследования. Разделение между рецессивным и доминантным типами не является «жестким и быстрым», хотя разделение между аутосомным и Х-сцепленным типами является (поскольку последние типы различаются исключительно на основе хромосомного расположения гена). Например, распространенная форма карликовости , ахондроплазия , обычно считается доминантным заболеванием, но дети с двумя генами ахондроплазии имеют тяжелое и обычно смертельное заболевание скелета, носителем которого можно считать ахондроплазию. Серповидно-клеточная анемия также считается рецессивным заболеванием, но гетерозиготные носители имеют повышенную устойчивость к малярии в раннем детстве, что можно охарактеризовать как родственное доминантное заболевание. [18] Когда пара, у которой один партнер или оба поражены заболеванием или являются носителями моногенного заболевания, желает иметь ребенка, они могут сделать это посредством экстракорпорального оплодотворения, которое позволяет провести преимплантационную генетическую диагностику, чтобы проверить, есть ли у эмбриона генетическое заболевание. [19]
Большинство врожденных нарушений обмена веществ , известных как врожденные нарушения обмена веществ, возникают в результате дефектов одного гена. Многие такие дефекты одного гена могут снизить приспособленность затронутых людей и, следовательно, присутствовать в популяции с более низкой частотой по сравнению с тем, что можно было бы ожидать на основе простых вероятностных расчетов. [20]
Аутосомно-доминантный
[ редактировать ]Для того чтобы у человека развилось аутосомно-доминантное заболевание, достаточно одной мутировавшей копии гена. У каждого пострадавшего человека обычно есть один затронутый родитель. [21] : 57 Вероятность того, что ребенок унаследует мутировавший ген, составляет 50%. Аутосомно-доминантные состояния иногда имеют пониженную пенетрантность . Это означает, что, хотя необходима только одна мутировавшая копия, не у всех людей, унаследовавших эту мутацию, развивается заболевание. Примерами этого типа расстройств являются болезнь Гентингтона , [21] : 58 нейрофиброматоз 1-го типа , нейрофиброматоз 2-го типа , синдром Марфана , наследственный неполипозный колоректальный рак , наследственные множественные экзостозы (высокопроникающее аутосомно-доминантное заболевание), туберозный склероз , болезнь фон Виллебранда и острая перемежающаяся порфирия . Врожденные дефекты также называют врожденными аномалиями. [22]
Аутосомно-рецессивный
[ редактировать ]Две копии гена должны быть мутированы, чтобы человек заболел аутосомно-рецессивным заболеванием. У пораженного человека обычно есть здоровые родители, каждый из которых несет одну копию мутировавшего гена и называется генетическим носителем . Каждый родитель с дефектным геном обычно не имеет симптомов. [23] Два здоровых человека, каждый из которых является носителем одной копии мутировавшего гена, имеют 25%-ный риск рождения ребенка с этим заболеванием при каждой беременности. Примерами этого типа расстройств являются альбинизм , дефицит ацил-КоА-дегидрогеназы средней цепи , муковисцидоз , серповидноклеточная анемия , болезнь Тея-Сакса , болезнь Нимана-Пика , спинальная мышечная атрофия и синдром Робертса . Некоторые другие фенотипы, такие как влажная или сухая ушная сера , также определяются по аутосомно-рецессивному типу. [24] [25] Некоторые аутосомно-рецессивные заболевания распространены потому, что в прошлом носительство одного из дефектных генов приводило к незначительной защите от инфекционных заболеваний или токсинов , таких как туберкулез или малярия . [26] К таким заболеваниям относятся муковисцидоз, [27] серповидно-клеточная анемия, [28] фенилкетонурия [29] и талассемия . [30]
 Наследственные дефекты ферментов обычно наследуются аутосомно, поскольку не-Х-хромосом больше, чем Х-хромосом, и рецессивно, поскольку ферментов непораженных генов обычно достаточно для предотвращения симптомов у носителей.
Наследственные дефекты ферментов обычно наследуются аутосомно, поскольку не-Х-хромосом больше, чем Х-хромосом, и рецессивно, поскольку ферментов непораженных генов обычно достаточно для предотвращения симптомов у носителей.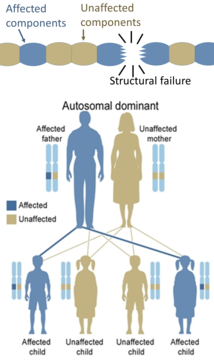 С другой стороны, наследственные дефекты структурных белков (такие как несовершенный остеогенез , синдром Марфана и многие синдромы Элерса-Данлоса ) обычно являются аутосомно-доминантными, поскольку дефектов некоторых компонентов достаточно, чтобы сделать всю структуру дисфункциональной. Это доминантно-негативный процесс, при котором мутированный генный продукт отрицательно влияет на немутантный генный продукт в той же клетке.
С другой стороны, наследственные дефекты структурных белков (такие как несовершенный остеогенез , синдром Марфана и многие синдромы Элерса-Данлоса ) обычно являются аутосомно-доминантными, поскольку дефектов некоторых компонентов достаточно, чтобы сделать всю структуру дисфункциональной. Это доминантно-негативный процесс, при котором мутированный генный продукт отрицательно влияет на немутантный генный продукт в той же клетке.


Х-сцепленная доминанта
[ редактировать ]
Х-сцепленные доминантные расстройства вызваны мутациями в генах Х-хромосомы . Лишь немногие заболевания имеют такой тип наследования, ярким примером является Х-сцепленный гипофосфатемический рахит . Этим расстройствам страдают как мужчины, так и женщины, при этом мужчины обычно страдают более серьезно, чем женщины. Некоторые Х-сцепленные доминантные состояния, такие как синдром Ретта , пигментное недержание 2-го типа и синдром Айкарди , обычно приводят к летальному исходу у мужчин либо внутриутробно , либо вскоре после рождения, и поэтому преимущественно наблюдаются у женщин. Исключением из этого вывода являются чрезвычайно редкие случаи, когда мальчики с синдромом Клайнфельтера (44+xxy) также наследуют Х-сцепленное доминантное заболевание и проявляют симптомы, более сходные с симптомами у женщин с точки зрения тяжести заболевания. Вероятность передачи Х-сцепленного доминантного расстройства у мужчин и женщин различна. Все сыновья мужчины с Х-сцепленным доминантным заболеванием не пострадают (поскольку они получают Y-хромосому своего отца), но все его дочери унаследуют это заболевание. У женщины с Х-сцепленным доминантным заболеванием вероятность рождения пораженного плода при каждой беременности составляет 50%, хотя в таких случаях, как пигментное недержание мочи, обычно жизнеспособны только потомки женского пола.
Х-сцепленный рецессивный
[ редактировать ]Х-сцепленные рецессивные состояния также вызываются мутациями в генах Х-хромосомы. Мужчины болеют гораздо чаще, чем женщины, поскольку у них есть только одна Х-хромосома, необходимая для проявления заболевания. Вероятность передачи расстройства различается у мужчин и женщин. Сыновья мужчины с Х-сцепленным рецессивным заболеванием не пострадают (поскольку они получают Y-хромосому своего отца), но его дочери будут носителями одной копии мутировавшего гена. Женщина-носитель Х-сцепленного рецессивного заболевания (Х Р Х р ) имеет 50%-ную вероятность рождения сыновей, пораженных этим заболеванием, и 50%-ную вероятность рождения дочерей, являющихся носителями одной копии мутировавшего гена. Х-сцепленные рецессивные состояния включают серьезные заболевания гемофилию А , мышечную дистрофию Дюшенна и синдром Леша-Нихана , а также распространенные и менее серьезные заболевания, такие как облысение по мужскому типу и красно-зеленую дальтонизм . Х-сцепленные рецессивные состояния иногда могут проявляться у женщин из-за искаженной Х-инактивации или моносомии Х ( синдром Тернера ). [ нужна ссылка ]
Y-связанный
[ редактировать ]Y-сцепленные расстройства вызваны мутациями Y-хромосомы. Эти состояния могут передаваться только от гетерогаметного пола (например, мужчины) потомству того же пола. Проще говоря, это означает, что Y-сцепленные расстройства у людей могут передаваться только от мужчин к их сыновьям; самки никогда не могут быть затронуты, поскольку у них нет Y-аллосом. [ нужна ссылка ]
Y-сцепленные расстройства встречаются чрезвычайно редко, но наиболее известные примеры обычно вызывают бесплодие. Репродукция в таких условиях возможна только путем обхода бесплодия путем медицинского вмешательства.
Митохондриальный
[ редактировать ]Этот тип наследования, также известный как материнское наследование, является самым редким и распространяется на 13 генов, кодируемых митохондриальной ДНК . Поскольку только яйцеклетки вносят митохондрии в развивающийся эмбрион, только матери (которые поражены) могут передать состояния митохондриальной ДНК своим детям. Примером этого типа расстройства является наследственная нейропатия зрительного нерва Лебера . [31]
Важно подчеркнуть, что подавляющее большинство митохондриальных заболеваний (особенно когда симптомы развиваются в раннем возрасте) на самом деле вызваны дефектом ядерного гена , поскольку митохондрии в основном развиваются из немитохондриальной ДНК. Эти заболевания чаще всего наследуются по аутосомно-рецессивному типу. [32]
Многофакторное расстройство
[ редактировать ]Генетические нарушения также могут быть сложными, многофакторными или полигенными, то есть они, вероятно, связаны с воздействием нескольких генов в сочетании с образом жизни и факторами окружающей среды. Многофакторные расстройства включают болезни сердца и диабет . Хотя сложные расстройства часто группируются в семьях, они не имеют четкой картины наследования. Это затрудняет определение риска унаследования или передачи этих заболеваний для человека. Сложные расстройства также трудно изучать и лечить, поскольку конкретные факторы, вызывающие большинство этих расстройств, еще не идентифицированы. Исследования, направленные на выявление причины сложных нарушений, могут использовать несколько методических подходов для определения генотип - фенотипических ассоциаций. Один из методов, подход «сначала генотип» , начинается с выявления генетических вариантов у пациентов, а затем определения связанных с ними клинических проявлений. Это противоречит более традиционному подходу, основанному на фенотипе, и может выявить причинные факторы, которые ранее были скрыты клиническими исследованиями. гетерогенность , пенетрантность и экспрессивность. [ нужна ссылка ]
В родословной полигенные заболевания имеют тенденцию «передаваться по наследству», но наследование не соответствует простым закономерностям, как в случае с менделевскими болезнями. Это не означает, что гены в конечном итоге невозможно обнаружить и изучить. Многие из них также имеют сильную экологическую составляющую (например, кровяное давление ).
Другие подобные случаи включают в себя:
- астма
- аутоиммунные заболевания, такие как рассеянный склероз
- рак
- цилиопатии
- волчья пасть
- диабет
- сердечное заболевание
- гипертония
- воспалительное заболевание кишечника
- умственная отсталость
- расстройство настроения
- ожирение
- ошибка рефракции
- бесплодие
Хромосомное заболевание
[ редактировать ]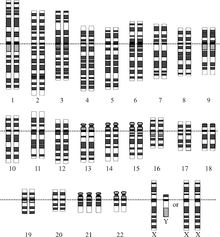
Хромосомное заболевание — это отсутствующая, дополнительная или неправильная часть хромосомной ДНК. [33] Это может быть связано с атипичным числом хромосом или структурной аномалией в одной или нескольких хромосомах. Примером таких нарушений является трисомия 21 (наиболее распространенная форма синдрома Дауна ), при которой во всех клетках имеется дополнительная копия хромосомы 21. [34]
Диагностика
[ редактировать ]Из-за широкого спектра известных генетических нарушений диагнозы широко варьируются и зависят от заболевания. Большинство генетических нарушений диагностируются до рождения , при рождении или в раннем детстве, однако некоторые из них, такие как болезнь Хантингтона , могут оставаться незамеченными до тех пор, пока у пациента не начнут проявляться симптомы во взрослом возрасте. [35]
Основные аспекты генетического заболевания основаны на наследственности генетического материала. Имея подробный семейный анамнез , можно предвидеть возможные заболевания у детей, что направляет медицинских работников на конкретные обследования в зависимости от расстройства и дает родителям возможность подготовиться к потенциальным изменениям образа жизни, предвидеть возможность мертворождения или рассмотреть возможность прерывания беременности . [36] Пренатальная диагностика может обнаружить наличие характерных отклонений в развитии плода с помощью ультразвука или обнаружить наличие характерных веществ с помощью инвазивных процедур , которые включают введение зондов или игл в матку, например, при амниоцентезе . [37]
Прогноз
[ редактировать ]Не все генетические нарушения напрямую приводят к смерти; однако не существует известных методов лечения генетических нарушений. Многие генетические нарушения влияют на стадии развития, например, синдром Дауна , тогда как другие приводят к чисто физическим симптомам, таким как мышечная дистрофия . Другие заболевания, такие как болезнь Хантингтона , не проявляют никаких признаков до взрослой жизни. В активное время генетического заболевания пациенты в основном полагаются на поддержание или замедление ухудшения качества жизни и сохранение автономии пациента . Это включает в себя физиотерапию и обезболивание .
Уход
[ редактировать ]
Лечение генетических нарушений — это непрекращающаяся битва: генной терапии . во всем мире уже завершено, продолжается или было одобрено более 1800 клинических испытаний [38] Несмотря на это, большинство вариантов лечения направлены на устранение симптомов расстройств с целью улучшить качество жизни пациентов .
Генная терапия относится к форме лечения, при которой пациенту вводится здоровый ген. Это должно облегчить дефект, вызванный дефектным геном, или замедлить прогрессирование заболевания. Основным препятствием была доставка генов в соответствующие клетки, ткани и органы, пораженные заболеванием. Исследователи изучили, как можно внедрить ген в потенциально триллионы клеток, несущих дефектную копию. Поиск ответа на этот вопрос стал препятствием между пониманием генетического заболевания и его коррекцией. [39]
Эпидемиология
[ редактировать ]Примерно 1 из 50 человек страдает известным моногенным заболеванием, а около 1 из 263 страдает хромосомным заболеванием . [7] Около 65% людей имеют те или иные проблемы со здоровьем в результате врожденных генетических мутаций. [7] Из-за значительного количества генетических нарушений примерно 1 из 21 человека страдает генетическим заболеванием, классифицируемым как « редкое » (обычно затрагивающее менее 1 из 2000 человек). Большинство генетических нарушений сами по себе редки. [5] [8] Известно более 6000 генетических нарушений. [4] в медицинской литературе постоянно описываются новые генетические нарушения. [5]
История
[ редактировать ]Самое раннее известное генетическое состояние гоминида было у ископаемых видов Paranthropusrobustus , причем более трети особей демонстрировали несовершенный амелогенез . [40]
См. также
[ редактировать ]- FINDbase (база данных частоты наследственных заболеваний)
- Генетическая эпидемиология
- Список генетических нарушений
- Группы населения в биомедицине
- Менделевская ошибка
Ссылки
[ редактировать ]- ^ «Генетические нарушения» . Узнайте.Генетика . Университет Юты. Архивировано из оригинала 15 июля 2022 г.
- ^ Львов Д., Фаворова О.О., Фаворов А.В. (июль 2012 г.). «Полигенный подход к изучению полигенных заболеваний» . Акта Натурэ . 4 (3): 59–71. дои : 10.32607/20758251-2012-4-3-59-71 . ПМЦ 3491892 . ПМИД 23150804 .
- ^ «Каковы различные способы наследования генетического заболевания?» . Домашний справочник по генетике . Архивировано из оригинала 27 сентября 2020 г. Проверено 14 января 2020 г.
- ^ Jump up to: а б «Статистика карты генов OMIM» . ОМИМ . Архивировано из оригинала 28 января 2020 г. Проверено 14 января 2020 г.
- ^ Jump up to: а б с д «О редких заболеваниях» . Сирота . Архивировано из оригинала 17 декабря 2019 г. Проверено 14 января 2020 г.
- ^ Бик Д., Бик С.Л., Диммок Д.П., Фаулер Т.А., Колфилд М.Дж., Скотт Р.Х. (март 2021 г.). «Онлайн-сборник излечимых генетических заболеваний» . Американский журнал медицинской генетики. Часть C. Семинары по медицинской генетике . 187 (1): 48–54. дои : 10.1002/ajmg.c.31874 . ПМЦ 7986124 . ПМИД 33350578 .
- ^ Jump up to: а б с д Кумар П., Радхакришнан Дж., Чоудхари М.А., Джампьетро П.Ф. (август 2001 г.). «Распространенность и закономерности проявления генетических нарушений в педиатрическом отделении неотложной помощи». Труды клиники Мэйо . 76 (8): 777–783. дои : 10.4065/76.8.777 . ПМИД 11499815 .
- ^ Jump up to: а б Джексон М., Маркс Л., Мэй Г.Х., Уилсон Дж.Б. (декабрь 2018 г.). «Генетическая основа болезней» . Очерки по биохимии . 62 (5): 643–723. дои : 10.1042/EBC20170053 . ПМК 6279436 . ПМИД 30509934 .
(рассчитано на основе «1 из 17» редких заболеваний и «80%» редких заболеваний, являющихся генетическими)
- ^ Хант Джей Ди. «Введение в рак» . Генетика и семьи Луизианы . lsuhsc.edu. Архивировано из оригинала 16 января 2020 года.
- ^ «Распространенность и заболеваемость редкими заболеваниями» (PDF) . Архивировано (PDF) из оригинала 18 ноября 2008 г.
- ^ «Запись OMIM № 144010 – ГИПЕРХОЛЕСТЕРОЛЕМИЯ, СЕМЕЙНАЯ, 2; FCHL2» . omim.org . Архивировано из оригинала 09 марта 2021 г. Проверено 1 июля 2019 г.
- ^ Джонсон Н.Е., Баттерфилд Р.Дж., Мейн К., Ньюкомб Т., Имбургия С., Данн Д. и др. (февраль 2021 г.). «Популяционная распространенность миотонической дистрофии типа 1 с использованием генетического анализа общегосударственной программы скрининга крови» . Неврология . 96 (7): е1045–е1053. дои : 10.1212/WNL.0000000000011425 . ПМК 8055332 . ПМИД 33472919 .
- ^ «Запись OMIM № 162200 – НЕЙРОФИБРОМАТОЗ, ТИП I; NF1» . omim.org . Архивировано из оригинала 8 марта 2021 г. Проверено 1 июля 2019 г.
- ^ Кин М.Г., Пьеритц Р.Э. (май 2008 г.). «Медицинское лечение синдрома Марфана». Тираж . 117 (21): 2802–2813. doi : 10.1161/CIRCULATIONAHA.107.693523 . ПМИД 18506019 .
- ^ Уокер Ф.О. (январь 2007 г.). «Болезнь Хантингтона». Ланцет . 369 (9557): 218–228. дои : 10.1016/S0140-6736(07)60111-1 . ПМИД 17240289 . S2CID 46151626 .
- ^ «Запись OMIM № 603903 – Серповидноклеточная АНЕМИЯ» . omim.org . Архивировано из оригинала 26 апреля 2021 г. Проверено 1 июля 2019 г.
- ^ Суонсон К. (ноябрь 2021 г.). «Аутосомно-рецессивный поликистоз почек» . Американский журнал акушерства и гинекологии . 225 (5). Эльзевир Б.В.: B7–B8. дои : 10.1016/j.ajog.2021.06.038 . ПМИД 34507795 . S2CID 237480065 .
- ^ Уильямс, Теннесси, Обаро, СК (июль 2011 г.). «Серповидноклеточная анемия и заболеваемость малярией: история с двумя хвостами». Тенденции в паразитологии . 27 (7): 315–320. дои : 10.1016/j.pt.2011.02.004 . ПМИД 21429801 .
- ^ Кулиев А, Верлинский Ю (апрель 2005 г.). «Преимплантационная диагностика: реалистичный вариант вспомогательной репродукции и генетической практики». Современное мнение в области акушерства и гинекологии . 17 (2): 179–183. дои : 10.1097/01.gco.0000162189.76349.c5 . ПМИД 15758612 . S2CID 9382420 .
- ^ Шимчикова Д., Хенеберг П. (декабрь 2019 г.). «Уточнение прогнозов эволюционной медицины на основе клинических данных о проявлениях менделевских болезней» . Научные отчеты . 9 (1): 18577. Бибкод : 2019NatSR...918577S . дои : 10.1038/s41598-019-54976-4 . ПМК 6901466 . ПМИД 31819097 .
- ^ Jump up to: а б Гриффитс А.Дж., Весслер С.Р., Кэрролл С.Б., Добли Дж. (2012). «2: Одногенное наследование». Введение в генетический анализ (10-е изд.). Нью-Йорк: WH Freeman and Company. ISBN 978-1-4292-2943-2 .
- ^ Малерб Х.Л., Модель Б, Бленкоу Х., Стронг К.Л., Олдос К. (июнь 2023 г.). «Обзор ключевой терминологии и определений, используемых для обозначения врожденных дефектов во всем мире» . Журнал общественной генетики . 14 (3): 241–262. дои : 10.1007/s12687-023-00642-2 . ПМЦ 10272040 . ПМИД 37093545 .
- ^ «Модели наследования одногенных заболеваний» . Learn.genetics.utah.edu . Архивировано из оригинала 1 июля 2019 г. Проверено 1 июля 2019 г.
- ^ Уэйд Н. (29 января 2006 г.). «Японские учёные идентифицировали ген ушной серы» . Нью-Йорк Таймс . Архивировано из оригинала 21 марта 2023 года . Проверено 20 февраля 2023 г.
- ^ Ёсиура К., Киносита А., Исида Т., Ниноката А., Исикава Т., Канаме Т. и др. (март 2006 г.). «SNP в гене ABCC11 является определяющим фактором типа ушной серы человека». Природная генетика . 38 (3): 324–330. дои : 10.1038/ng1733 . ПМИД 16444273 . S2CID 3201966 .
- ^ Миттон Дж. Б. (2002). «Гетерозиготное преимущество». ЭЛС . дои : 10.1038/npg.els.0001760 . ISBN 978-0-470-01617-6 .
- ^ Пулман Э.М., Гальвани А.П. (февраль 2007 г.). «Оценка кандидатов в средства селективного давления при муковисцидозе» . Журнал Королевского общества, Интерфейс . 4 (12): 91–98. дои : 10.1098/rsif.2006.0154 . ПМЦ 2358959 . ПМИД 17015291 .
- ^ Эллисон AC (октябрь 2009 г.). «Генетический контроль устойчивости человека к малярии». Современное мнение в иммунологии . 21 (5): 499–505. дои : 10.1016/j.coi.2009.04.001 . ПМИД 19442502 .
- ^ Вульф Л.И. (май 1986 г.). «Преимущество гетерозигот при фенилкетонурии» . Американский журнал генетики человека . 38 (5): 773–775. ПМК 1684820 . ПМИД 3717163 .
- ^ Уэзералл диджей (2015). «Талассемия: нарушения синтеза глобина» . Гематология Уильямса (9е изд.). МакГроу Хилл Профессионал. п. 725. ИСБН 978-0-07-183301-1 . Архивировано из оригинала 20 февраля 2023 г. Проверено 20 февраля 2023 г.
- ^ Шемеш А., Суд Г., Марголин Э. «Наследственная оптическая невропатия Лебера (LHON)» . StatPearls [Интернет] . Остров сокровищ (Флорида): StatPearls Publishing.
- ^ Нуссбаум Р., Макиннес Р., Уиллард Х (2007). Томпсон и Томпсон Генетика в медицине . Филадельфия, Пенсильвания: Сондерс. стр. 144, 145, 146. ISBN. 978-1-4160-3080-5 .
- ^ «Генетические заболевания: что это такое, типы, симптомы и причины» . Кливлендская клиника . Архивировано из оригинала 1 ноября 2023 г. Проверено 1 ноября 2023 г.
- ^ Центры по контролю и профилактике заболеваний (10 октября 2023 г.). «Факты о синдроме Дауна | CDC» . Центры по контролю и профилактике заболеваний . Архивировано из оригинала 28 июля 2017 г. Проверено 1 ноября 2023 г.
- ^ Вайант К.Дж., Риддер А.Дж., Даялу П. (апрель 2017 г.). «Обновленная информация о лечении болезни Хантингтона». Текущие отчеты по неврологии и нейробиологии . 17 (4): 33. дои : 10.1007/s11910-017-0739-9 . ПМИД 28324302 .
- ^ Милунский А, Милунский Дж. М. (2021). «Генетическое консультирование: предвзятое мнение, пренатальное и перинатальное». Генетические нарушения и плод . стр. 1–101. дои : 10.1002/9781119676980.ch1 . ISBN 978-1-119-67698-0 .
- ^ «Диагностические тесты – амниоцентез» . Гарвардская медицинская школа. Архивировано из оригинала 16 мая 2008 г. Проверено 15 июля 2008 г.
- ^ Гинн С.Л., Александр И.Е., Эдельштейн М.Л., Абеди М.Р., Виксон Дж. (февраль 2013 г.). «Клинические испытания генной терапии во всем мире до 2012 г. - обновленная информация». Журнал генной медицины . 15 (2): 65–77. дои : 10.1002/jgm.2698 . ПМИД 23355455 . S2CID 37123019 .
- ^ Верма И.М. (август 2013 г.). «Медицина. Генная терапия, которая работает». Наука . 341 (6148): 853–855. Бибкод : 2013Sci...341..853V . дои : 10.1126/science.1242551 . ПМИД 23970689 . S2CID 206550787 .
- ^ Таул I, ирландский доктор юридических наук (апрель 2019 г.). «Вероятное генетическое происхождение точечной гипоплазии эмали на коренных зубах Paranthropusrobustus» (PDF) . Журнал эволюции человека . 129 : 54–61. дои : 10.1016/j.jhevol.2019.01.002 . ПМИД 30904040 . S2CID 85502058 . Архивировано (PDF) из оригинала 4 июня 2023 г. Проверено 20 февраля 2023 г.
Внешние ссылки
[ редактировать ]- Геномика общественного здравоохранения в CDC
- OMIM - Интернет-менделевское наследование у человека, каталог человеческих генов и генетических нарушений.
- Информационный центр по генетическим и редким заболеваниям (GARD), Управление редких заболеваний (ORD), Национальные институты здравоохранения (NIH)
- Национальный центр CDC по врожденным дефектам и нарушениям развития
- Информация о генетических заболеваниях из проекта «Геном человека»
- Глобальный проект генов, Организация по генетическим и редким заболеваниям
- Список генетических заболеваний - Genome.gov