Химическая реакция

Химическая реакция – это процесс, приводящий к химическому превращению одного набора химических веществ в другой. [1] Когда происходят химические реакции, атомы перестраиваются, и реакция сопровождается изменением энергии по мере образования новых продуктов. Классически химические реакции включают в себя изменения, которые затрагивают только положения электронов при образовании и разрыве химических связей между атомами , без изменений в ядрах (без изменений в присутствующих элементах) и часто могут быть описаны химическим уравнением . Ядерная химия — это раздел химии, который включает химические реакции нестабильных и радиоактивных элементов , в которых могут происходить как электронные, так и ядерные изменения.
Вещество (или вещества), первоначально участвующие в химической реакции, называются реагентами или реагентами . Химические реакции обычно характеризуются химическими изменениями и приводят к образованию одного или нескольких продуктов , свойства которых обычно отличаются от свойств реагентов. Реакции часто состоят из последовательности отдельных подэтапов, так называемых элементарных реакций , и информация о точном ходе действия является частью механизма реакции . Химические реакции описываются химическими уравнениями , которые символически представляют исходные вещества, конечные продукты, а иногда и промежуточные продукты и условия реакции.
Химические реакции происходят с характерной скоростью реакции при заданной температуре и химической концентрации. Некоторые реакции производят тепло и называются экзотермическими реакциями , в то время как другие могут требовать тепла для протекания реакции, которые называются эндотермическими реакциями . Обычно скорость реакции увеличивается с повышением температуры, поскольку имеется больше тепловой энергии, необходимой для достижения энергии активации, необходимой для разрыва связей между атомами.
Реакцию можно классифицировать как окислительно-восстановительную, при которой происходят окисление и восстановление , или неокислительно-восстановительную, при которой окисление и восстановление не происходят. Большинство простых окислительно-восстановительных реакций можно классифицировать как реакции сочетания, разложения или одиночного замещения.
используются различные химические реакции При химическом синтезе для получения желаемого продукта. В биохимии последовательная серия химических реакций (где продукт одной реакции является реагентом следующей реакции) образует метаболические пути . Эти реакции часто катализируются белковыми ферментами . Ферменты увеличивают скорость биохимических реакций, так что метаболический синтез и разложение, невозможные в обычных условиях, могут происходить при температуре и концентрациях, присутствующих внутри клетки .
Общая концепция химической реакции была расширена на реакции между объектами, меньшими, чем атомы, включая ядерные реакции , радиоактивные распады и реакции между элементарными частицами , как описано квантовой теорией поля .
История

Химические реакции, такие как горение в огне, брожение и восстановление руд до металлов, были известны с древности. Первоначальные теории преобразования материалов были разработаны греческими философами, например, « Теория четырех элементов» Эмпедокла , утверждающая, что любое вещество состоит из четырех основных элементов — огня, воды, воздуха и земли. В средние века химические превращения изучали алхимики . Они пытались, в частности, превратить свинец в золото , для чего использовали реакции свинца и свинцово-медных сплавов с серой . [2]
Искусственное производство химических веществ уже было главной целью средневековых алхимиков. [3] Примеры включают синтез хлорида аммония из органических веществ , как описано в работах (ок. 850–950), приписываемых Джабиру ибн Хайяну , [4] или производство минеральных кислот , таких как серная и азотная кислоты , более поздними алхимиками, начиная с ок. 1300. [5] Производство минеральных кислот включало нагревание сульфатных и нитратных минералов, таких как медный купорос , квасцы и селитра . В 17 веке Иоганн Рудольф Глаубер получил соляную кислоту и сульфат натрия путем взаимодействия серной кислоты и хлорида натрия . С разработкой свинцово-камерного процесса в 1746 году и процесса Леблана , позволившего крупномасштабное производство серной кислоты и карбоната натрия соответственно, химические реакции стали внедряться в промышленность. Дальнейшая оптимизация технологии серной кислоты привела к контактному процессу в 1880-х годах. [6] а процесс Габера был разработан в 1909–1910 годах для синтеза аммиака . [7]
С 16-го века исследователи, в том числе Ян Баптист ван Гельмонт , Роберт Бойль и Исаак Ньютон, пытались создать теории экспериментально наблюдаемых химических превращений. Теория флогистона была предложена в 1667 году Иоганном Иоахимом Бехером . Он постулировал существование огнеподобного элемента под названием «флогистон», который содержится в горючих телах и выделяется при горении . Это оказалось ложным в 1785 году Антуаном Лавуазье , который нашел правильное объяснение горения реакцией с кислородом воздуха. [8]
Жозеф Луи Гей-Люссак в 1808 году признал, что газы всегда реагируют в определенных отношениях друг с другом. На основе этой идеи и атомной теории Джона Дальтона Ж. Пруст разработал закон определенных пропорций , который впоследствии привел к появлению представлений о стехиометрии и химических уравнениях . [9]
Что касается органической химии , то долгое время считалось, что соединения, полученные из живых организмов, слишком сложны, чтобы их можно было получить синтетическим путем . Согласно концепции витализма , органическое вещество наделялось «жизненной силой» и отличалось от неорганических материалов. Однако это разделение было прекращено синтезом мочевины из неорганических предшественников Фридрихом Вёлером в 1828 году. Среди других химиков, внесших большой вклад в органическую химию, - Александр Уильям Уильямсон с его синтезом простых эфиров и Кристофер Кельк Ингольд , который, среди многих открытий, установил механизмы реакций замещения .
Характеристики
Этот раздел нуждается в расширении . Вы можете помочь, добавив к нему . ( ноябрь 2020 г. ) |
Общими характеристиками химических реакций являются:
- Эволюция газа
- Образование осадка
- Изменение температуры
- Изменение состояния
Уравнения
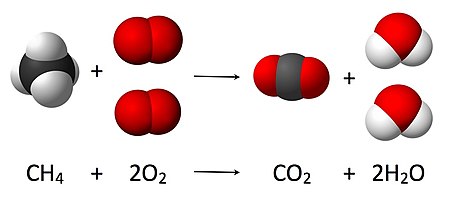
Химические уравнения используются для графической иллюстрации химических реакций. Они состоят из химических или структурных формул реагентов слева и продуктов справа. Они разделены стрелкой (→), указывающей направление и тип реакции; стрелка читается как слово «уступает». [10] Кончик стрелки указывает направление, в котором протекает реакция. Двойная стрелка (⇌), указывающая в противоположные стороны, используется для равновесных реакций . Уравнения должны быть сбалансированы по стехиометрии , количество атомов каждого вида должно быть одинаковым в обеих частях уравнения. Это достигается путем масштабирования количества участвующих молекул (A, B, C и D в схематическом примере ниже) на соответствующие целые числа a, b, c и d . [11]
- а А + б В → с С + d D
Более сложные реакции представлены схемами реакций, на которых помимо исходных веществ и продуктов показаны важные промежуточные или переходные состояния . Кроме того, над стрелкой реакции могут быть указаны некоторые относительно незначительные добавки к реакции; примерами таких дополнений являются вода, тепло, освещение, катализатор и т. д. Аналогичным образом, некоторые второстепенные продукты могут быть помещены под стрелкой, часто со знаком минус.
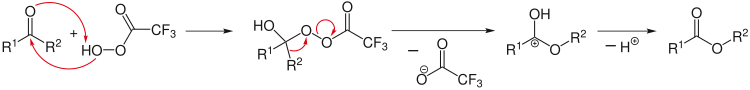
Ретросинтетический анализ можно применять для разработки сложной реакции синтеза. Здесь анализ начинается с продуктов, например, путем расщепления выбранных химических связей, чтобы получить возможные исходные реагенты. В ретро-реакциях используется специальная стрелка (⇒). [12]
Элементарные реакции
Элементарная реакция — наименьшее деление, на которое можно разложить химическую реакцию, она не имеет промежуточных продуктов. [13] Большинство экспериментально наблюдаемых реакций состоят из множества элементарных реакций, протекающих параллельно или последовательно. Реальная последовательность отдельных элементарных реакций известна как механизм реакции . В элементарной реакции участвует несколько молекул, обычно одна или две, поскольку вероятность встречи нескольких молекул в определенное время мала. [14]

Важнейшими элементарными реакциями являются мономолекулярные и бимолекулярные реакции. В мономолекулярной реакции участвует только одна молекула; он преобразуется путем изомеризации или диссоциации в одну или несколько других молекул. Такие реакции требуют добавления энергии в виде тепла или света. Типичным примером мономолекулярной реакции является цис-транс -изомеризация , при которой цис-форма соединения превращается в транс-форму или наоборот. [15]
В типичной реакции диссоциации связь в молекуле расщепляется ( рвется ), в результате чего образуются два молекулярных фрагмента. Расщепление может быть гомолитическим или гетеролитическим . В первом случае связь разделяется так, что каждый продукт сохраняет электрон и становится нейтральным радикалом . Во втором случае оба электрона химической связи остаются у одного из продуктов, в результате чего образуются заряженные ионы . Диссоциация играет важную роль в запуске цепных реакций , таких как водородно-кислородные реакции или реакции полимеризации .
- Диссоциация молекулы АВ на фрагменты А и В

В бимолекулярных реакциях две молекулы сталкиваются и реагируют друг с другом. Их слияние называется химическим синтезом или реакцией присоединения .

Другая возможность состоит в том, что только часть одной молекулы передается другой молекуле. Реакции такого типа происходят, например, в окислительно-восстановительных и кислотно-основных реакциях. В окислительно-восстановительных реакциях переносимой частицей является электрон, тогда как в кислотно-основных реакциях — протон. Этот тип реакции еще называют метатезисом .

например

Химическое равновесие
Большинство химических реакций обратимы; то есть они могут бежать и бегут в обоих направлениях. Прямая и обратная реакции конкурируют друг с другом и различаются по скорости реакции . Эти скорости зависят от концентрации и поэтому изменяются со временем реакции: обратная скорость постепенно увеличивается и становится равной скорости прямой реакции, устанавливая так называемое химическое равновесие. Время достижения равновесия зависит от таких параметров, как температура, давление и используемые материалы, и определяется минимальной свободной энергией . В равновесии свободная энергия Гиббса реакции должна быть равна нулю. Зависимость от давления можно объяснить принципом Ле Шателье . Например, увеличение давления из-за уменьшения объема приводит к смещению реакции в сторону с меньшим количеством молей газа. [16]
Выход реакции стабилизируется при равновесии, но его можно увеличить, удаляя продукт из реакционной смеси, или изменить, увеличив температуру или давление. Изменение концентраций реагентов не влияет на константу равновесия, но влияет на положение равновесия.
Термодинамика
Химические реакции определяются законами термодинамики . Реакции могут протекать сами по себе, если они экзергоничны , то есть выделяют свободную энергию. Соответствующее изменение свободной энергии реакции состоит из изменений двух различных термодинамических величин, энтальпии и энтропии : [17]
- .
- G : свободная энергия, H : энтальпия, T : температура, S : энтропия, Δ : разница (изменение между оригиналом и продуктом)

Реакции могут быть экзотермическими , где ΔH отрицательна и выделяется энергия. Типичными примерами экзотермических реакций являются горение , осаждение и кристаллизация , при которых упорядоченные твердые вещества образуются из неупорядоченных газовых или жидких фаз. Напротив, в эндотермических реакциях тепло потребляется из окружающей среды. Это может происходить за счет увеличения энтропии системы, часто за счет образования газообразных или растворенных продуктов реакции, имеющих более высокую энтропию. Поскольку энтропийный член изменения свободной энергии увеличивается с температурой, многие эндотермические реакции предпочтительно протекают при высоких температурах. Напротив, многие экзотермические реакции, такие как кристаллизация, протекают предпочтительно при более низких температурах. Изменение температуры иногда может изменить знак энтальпии реакции, как при оксидом углерода диоксида восстановлении молибдена :
- ;


Эта реакция с образованием углекислого газа и молибдена является эндотермической при низких температурах и становится менее эндотермической с повышением температуры. [18] ΔH , и выше этой ° равна нулю при 1855 К температуры реакция становится экзотермической.
Изменения температуры также могут изменить направление реакции. Например, реакция конверсии водяного газа

этому благоприятствуют низкие температуры, а обратному - высокие температуры. Смена направления реакции происходит при 1100 К. [18]
Реакции также можно характеризовать изменением их внутренней энергии , учитывающей изменения энтропии, объема и химического потенциала . Последнее зависит, в том числе, от активности участвующих веществ. [19]
- U : внутренняя энергия, S : энтропия, p : давление, µ : химический потенциал, n : количество молекул, d : знак малого изменения.

Кинетика
Скорость протекания реакций изучается с помощью кинетики реакций . Ставка зависит от различных параметров, таких как:
- Концентрации реагентов , которые обычно ускоряют реакцию, если их повысить за счет увеличения количества столкновений в единицу времени. Однако скорость некоторых реакций не зависит от концентрации реагентов из-за ограниченного числа каталитических центров. Такие реакции называются реакциями нулевого порядка .
- Площадь поверхности, доступная для контакта между реагентами, в частности твердыми в гетерогенных системах. Большая площадь поверхности приводит к более высокой скорости реакции.
- Давление – увеличение давления уменьшает объем между молекулами и, следовательно, увеличивает частоту столкновений между молекулами.
- Энергия активации , которая определяется как количество энергии, необходимое для того, чтобы реакция началась и продолжилась самопроизвольно. Более высокая энергия активации означает, что реагентам для начала требуется больше энергии, чем реакции с более низкой энергией активации.
- Температура , которая ускоряет реакции, если ее повысить, поскольку более высокая температура увеличивает энергию молекул, создавая больше столкновений в единицу времени,
- Наличие или отсутствие катализатора . Катализаторы — это вещества, которые образуют слабые связи с реагентами или промежуточными продуктами и изменяют путь (механизм) реакции, что, в свою очередь, увеличивает скорость реакции за счет снижения энергии активации, необходимой для протекания реакции. Катализатор не разрушается и не изменяется в ходе реакции, поэтому его можно использовать повторно.
- Для некоторых реакций присутствие электромагнитного излучения , особенно ультрафиолетового света , необходимо, чтобы способствовать разрыву связей и начать реакцию. Это особенно справедливо для реакций с участием радикалов .
Несколько теорий позволяют рассчитывать скорости реакций на молекулярном уровне. Это поле называется динамикой реакции. Скорость v реакции первого порядка , которой может быть распад вещества А, определяется выражением:
![{\displaystyle v=-{\frac {d[{\ce {A}}]}{dt}}=k\cdot [{\ce {A}}].}](https://wikimedia.org/api/rest_v1/media/math/render/svg/12291760fcaff20a02ff74abd0dfcb922664cddb)
Его интеграция дает:
![{\displaystyle {\ce {[A]}}(t)={\ce {[A]}}_{0}\cdot e^{-k\cdot t}.}](https://wikimedia.org/api/rest_v1/media/math/render/svg/498c37558508e2f7297604f93bb5408dcd8c3fd4)
Здесь k — константа скорости первого порядка, имеющая размерность 1/время, [A]( t ) — концентрация в момент времени t , а [A] 0 — начальная концентрация. Скорость реакции первого порядка зависит только от концентрации и свойств участвующего вещества, а саму реакцию можно описать характерным периодом полураспада . При описании реакций более высокого порядка необходимо более одной постоянной времени. Температурная зависимость константы скорости обычно подчиняется уравнению Аррениуса :

где E a — энергия активации, а k B — постоянная Больцмана . Одной из простейших моделей скорости реакции является теория столкновений . Более реалистичные модели адаптированы к конкретной проблеме и включают теорию переходного состояния , расчет поверхности потенциальной энергии , теорию Маркуса и теорию Райса-Рамспергера-Касселя-Маркуса (РРКМ) . [20]
Типы реакций
Четыре основных типа
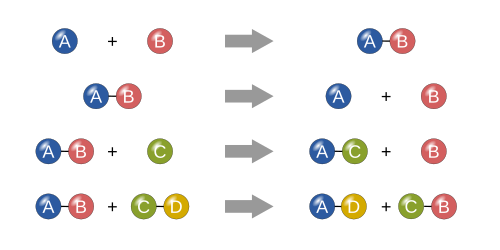
Синтез
В реакции синтеза два или более простых веществ соединяются с образованием более сложного вещества. Эти реакции имеют общий вид:

Два или более реагента, дающие один продукт, — это еще один способ идентификации реакции синтеза. Одним из примеров реакции синтеза является соединение железа и серы с образованием сульфида железа (II) :

Другой пример — простой газообразный водород в сочетании с простым газообразным кислородом для получения более сложного вещества, такого как вода. [21]
Разложение
Реакция разложения – это когда более сложное вещество распадается на более простые части. Таким образом, это противоположность реакции синтеза и может быть записано как [21]

Одним из примеров реакции разложения является электролиз воды с образованием кислорода и водорода :

Одиночное смещение
В одной реакции замещения один несвязанный элемент заменяет другой в соединении; другими словами, один элемент меняется местами с другим элементом в составе соединения. [21] Эти реакции имеют общую форму:

Одним из примеров реакции одиночного замещения является ситуация, когда магний заменяет водород в воде с образованием твердого гидроксида магния и газообразного водорода:

Двойное смещение
В реакции двойного замещения анионы и катионы двух соединений меняются местами и образуют два совершенно разных соединения. Эти реакции имеют общий вид: [21]

Например, при реакции хлорида бария (BaCl 2 ) и сульфата магния (MgSO 4 ) SO 4 2− анион меняется местами с 2Cl − анион, дающий соединения BaSO 4 и MgCl 2 .
Другим примером реакции двойного замещения является реакция нитрата свинца (II) с йодидом калия с образованием йодида свинца (II) и нитрата калия :

Прямые и обратные реакции
Согласно принципу Ле Шателье , реакции могут протекать в прямом или обратном направлении, пока не закончатся или не достигнут равновесия . [22]
Прямые реакции
Реакции, протекающие в прямом направлении до достижения равновесия, часто называют спонтанными реакциями , то есть является отрицательным, что означает, что если они происходят при постоянной температуре и давлении, они уменьшают свободную энергию Гиббса реакции. Им не требуется много энергии, чтобы двигаться вперед. [23] Большинство реакций являются прямыми реакциями. Примеры:

- Реакция водорода и кислорода с образованием воды.
- 2ч
2 + О
2 ⇌ 2Ч
22О
- Диссоциация уксусной кислоты в воде на ацетат- ионы и ионы гидроксония .
- СН
3 СООН + Н
2 О ⇌ СН
3 операционный директор −
+ Ч
33О +
Обратная реакция
Реакции, протекающие в обратном направлении для достижения равновесия, часто называют неспонтанными реакциями , т. е. положителен, что означает, что если они происходят при постоянной температуре и давлении, они увеличивают свободную энергию Гиббса реакции. Им требуется затрата энергии, чтобы двигаться вперед. [23] [24] Примеры включают в себя:
- Зарядка обычной батареи постоянного тока (состоящей из электролитических элементов ) от внешнего источника электроэнергии [25]
- Фотосинтез , обусловленный поглощением электромагнитного излучения , обычно в виде солнечного света. [26]
- + + → +
Горение
В реакции горения элемент или соединение вступает в реакцию с окислителем, обычно кислородом , часто выделяя энергию в виде тепла или света . В реакциях горения часто участвуют углеводороды . Например, сгорание 1 моля (114 г) октана в кислороде

выделяет 5500 кДж. Реакция горения также может возникнуть в результате реакции углерода , магния или серы с кислородом. [27]


Окисление и восстановление


Окислительно-восстановительные реакции можно понимать как перенос электронов от одного участвующего вещества ( восстановителя ) к другому ( окислителю ). В этом процессе первый вид окисляется , а второй восстанавливается . Хотя эти описания достаточны для многих целей, они не совсем верны. Окисление лучше определить как увеличение степени окисления атомов, а восстановление – как уменьшение степени окисления. На практике перенос электронов всегда меняет степень окисления, но существует множество реакций, которые классифицируются как «окислительно-восстановительные», даже если перенос электронов не происходит (например, с участием ковалентных связей). [28] [29]
В следующей окислительно-восстановительной реакции опасный металлический натрий реагирует с токсичным газообразным хлором с образованием ионного соединения хлорида натрия или обычной поваренной соли:

В ходе реакции металлический натрий переходит от степени окисления 0 (чистый элемент) к +1: другими словами, натрий потерял один электрон и считается, что он окислился. С другой стороны, газообразный хлор переходит от окисления 0 (также чистого элемента) к -1: хлор приобретает один электрон и, как говорят, восстанавливается. Поскольку хлор восстанавливается, он считается акцептором электронов или, другими словами, вызывает окисление натрия – таким образом, газообразный хлор считается окислителем. И наоборот, натрий окисляется или является донором электронов и, таким образом, вызывает восстановление других видов и считается восстановителем .
Какой из участвующих реагентов будет восстановителем или окислителем, можно предсказать по электроотрицательности их элементов. Элементы с низкой электроотрицательностью, такие как большинство металлов, легко отдают электроны и окисляются – они являются восстановителями. Напротив, многие оксиды или ионы с высокими степенями окисления некислородных атомов, такие как H
22О
2 , МnО −
4 , КрО
3 , Кр
22О 2−
7 , или ОсО
4 , могут получить один или два дополнительных электрона и являются сильными окислителями.
Для некоторых элементов основной группы количество электронов, отдаваемых или принимаемых в окислительно-восстановительной реакции, можно предсказать на основе электронной конфигурации реагирующего элемента. Элементы пытаются достичь конфигурации благородного газа с низкой энергией , поэтому щелочные металлы и галогены будут отдавать и принимать по одному электрону соответственно. Сами по себе благородные газы химически неактивны. [30]
Общую окислительно-восстановительную реакцию можно сбалансировать, объединив полуреакции окисления и восстановления, умноженные на коэффициенты так, чтобы количество электронов, потерянных при окислении, равнялось количеству электронов, полученных при восстановлении.
Важным классом окислительно-восстановительных реакций являются электролитические электрохимические реакции, в которых электроны от источника питания на отрицательном электроде используются в качестве восстановителя, а отвод электронов на положительном электроде - в качестве окислителя. Эти реакции особенно важны для производства химических элементов, таких как хлор. [31] или алюминий . Обратный процесс, при котором электроны высвобождаются в окислительно-восстановительных реакциях и химическая энергия преобразуется в электрическую, возможен и используется в батареях .
Комплексообразование
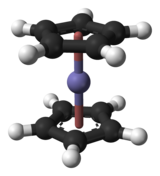
В реакциях комплексообразования несколько лигандов реагируют с атомом металла с образованием координационного комплекса . Это достигается за счет размещения неподеленных пар лиганда на пустых орбиталях атома металла и образования диполярных связей . Лиганды — основания Льюиса , они могут быть как ионами, так и нейтральными молекулами, например окисью углерода, аммиаком или водой. Число лигандов, которые реагируют с центральным атомом металла, можно найти с помощью правила 18 электронов , согласно которому валентные оболочки переходного металла коллективно размещают 18 электронов , тогда как симметрию образующегося комплекса можно предсказать с помощью кристаллического поля. теория и теория поля лигандов . Реакции комплексообразования также включают обмен лигандов , при котором один или несколько лигандов заменяются другими, и окислительно-восстановительные процессы, которые изменяют степень окисления центрального атома металла. [32]
Кислотно-основные реакции
В кислотно-основной теории Бренстеда-Лоури кислотно -основная реакция включает перенос протонов ( H + ) от одного вида ( кислоты ) к другому ( основанию ). Когда протон удаляется из кислоты, образующаяся разновидность называется основанием, сопряженным с этой кислотой . Когда протон принимается основанием, образующаяся разновидность называется кислотой, сопряженной с этим основанием . [33] Другими словами, кислоты действуют как доноры протонов, а основания действуют как акцепторы протонов в соответствии со следующим уравнением:

Возможна обратная реакция, поэтому кислота/основание и сопряженное основание/кислота всегда находятся в равновесии. Равновесие определяется константами диссоциации кислоты и основания ( K a и K b ) участвующих веществ. Особым случаем кислотно-основной реакции является нейтрализация , при которой кислота и основание, взятые в одинаковых количествах, образуют нейтральную соль .
Кислотно-основные реакции могут иметь разные определения в зависимости от используемой концепции кислотно-основной реакции. Некоторые из наиболее распространенных:
- Определение Аррениуса : Кислоты диссоциируют в воде с выделением H 3 O. + ионы; основания диссоциируют в воде с выделением ОН − ионы.
- Определение Бренстеда-Лоури : Кислоты представляют собой протон ( H + ) доноры, основания являются акцепторами протонов; это включает в себя определение Аррениуса.
- Определение Льюиса : Кислоты являются акцепторами электронных пар, а основания - донорами электронных пар; сюда входит определение Брёнстеда-Лоури.
Осадки

Осаждение – это образование твердого вещества в растворе или внутри другого твердого вещества в ходе химической реакции. Обычно это происходит, когда концентрация растворенных ионов превышает растворимости. предел [34] и образует нерастворимую соль. Этому процессу можно способствовать добавлением осаждающего агента или удалением растворителя. Быстрое осаждение приводит к образованию аморфного или микрокристаллического остатка, а медленный процесс может привести к образованию монокристаллов . Последний также может быть получен перекристаллизацией из микрокристаллических солей. [35]
Твердотельные реакции
Реакции могут происходить между двумя твердыми телами. Однако из-за относительно небольших скоростей диффузии в твердых телах соответствующие химические реакции протекают очень медленно по сравнению с реакциями в жидкой и газовой фазах. Их ускоряют за счет повышения температуры реакции и тонкого разделения реагента для увеличения площади контактирующей поверхности. [36]
Реакции на границе раздела твердое тело/газ
Реакция может происходить на границе твердого тела и газа, на поверхностях при очень низком давлении, например, в сверхвысоком вакууме . С помощью сканирующей туннельной микроскопии можно наблюдать реакции на границе твердого тела и газа в реальном пространстве, если временной масштаб реакции находится в правильном диапазоне. [37] [38] Реакции на границе твердого тела и газа в некоторых случаях связаны с катализом.
Фотохимические реакции
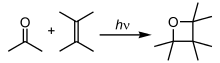
В фотохимических реакциях атомы и молекулы поглощают энергию ( фотоны ) освещающего света и переводят ее в возбужденное состояние . Затем они могут высвободить эту энергию, разрывая химические связи, создавая тем самым радикалы. Фотохимические реакции включают водородно-кислородные реакции, радикальную полимеризацию , цепные реакции и реакции перегруппировки . [39]
Многие важные процессы связаны с фотохимией. Ярким примером является фотосинтез , при котором большинство растений используют солнечную энергию для преобразования углекислого газа и воды в глюкозу , утилизируя кислород в качестве побочного продукта. Люди полагаются на фотохимию для образования витамина D, а зрение инициируется фотохимической реакцией родопсина . [15] У светлячков фермент к в брюшной полости катализирует реакцию, приводящую биолюминесценции . [40] Многие важные фотохимические реакции, такие как образование озона, происходят в атмосфере Земли и составляют химию атмосферы .
Катализ


При катализе реакция протекает не напрямую, а через реакцию с третьим веществом, известным как катализатор . Хотя катализатор участвует в реакции, образуя слабые связи с реагентами или интермедиатами, к концу реакции он возвращается в исходное состояние и поэтому не расходуется. Однако он может быть ингибирован, деактивирован или разрушен вторичными процессами. Катализаторы могут использоваться в другой фазе ( гетерогенные ) или в той же фазе ( гомогенные ), что и реагенты. В гетерогенном катализе типичные вторичные процессы включают коксование , при котором катализатор покрывается полимерными побочными продуктами. Кроме того, гетерогенные катализаторы могут растворяться в растворе в системе твердое тело-жидкость или испаряться в системе твердое тело-газ. Катализаторы могут только ускорить реакцию – химические вещества, замедляющие реакцию, называются ингибиторами. [41] [42] Вещества, повышающие активность катализаторов, называются промоторами, а вещества, дезактивирующие катализаторы, — каталитическими ядами. При использовании катализатора реакция, кинетически ингибируемая высокой энергией активации, может происходить в обход этой энергии активации.
Гетерогенные катализаторы обычно представляют собой твердые вещества, измельченные в порошок, чтобы максимизировать площадь их поверхности. Особое значение в гетерогенном катализе имеют металлы платиновой группы и другие переходные металлы, которые используются при гидрировании , каталитическом риформинге и в синтезе таких товарных химикатов, как азотная кислота и аммиак . Кислоты являются примером гомогенного катализатора, они повышают нуклеофильность карбонилов , позволяя протекать реакции, которые иначе не протекали бы с электрофилами. Преимущество гомогенных катализаторов заключается в простоте их смешивания с реагентами, но их также может быть трудно отделить от продуктов. Поэтому во многих промышленных процессах предпочтение отдается гетерогенным катализаторам. [43]
Реакции в органической химии
В органической химии , помимо реакций окисления, восстановления или кислотно-основных реакций, может протекать ряд других реакций, в которых участвуют ковалентные связи между атомами углерода или углеродом и гетероатомами (такими как кислород, азот, галогены и т. д.). Многие специфические реакции в органической химии носят название реакций, названных в честь их первооткрывателей.
Одной из наиболее важных в промышленном отношении реакций является крекинг тяжелых углеводородов на нефтеперерабатывающих заводах с целью создания более мелких и простых молекул. Этот процесс используется для производства бензина . Конкретные типы органических реакций можно сгруппировать по их механизмам реакции (в частности, замещению, присоединению и удалению) или по типам продуктов, которые они производят (например, метилирование , полимеризация и галогенирование ).
Замена
В реакции замещения функциональная группа в конкретном химическом соединении заменяется другой группой. [44] Эти реакции можно различать по типу замещающих частиц на нуклеофильное , электрофильное или радикальное замещение .

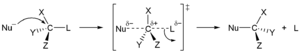
В первом типе нуклеофил , атом или молекула с избытком электронов и, следовательно, с отрицательным или частичным зарядом , заменяет другой атом или часть молекулы-субстрата. Электронная пара нуклеофила атакует подложку, образуя новую связь, а уходящая группа уходит с электронной парой. Нуклеофил может быть электрически нейтральным или отрицательно заряженным, тогда как субстрат обычно является нейтральным или положительно заряженным. Примерами нуклеофилов являются гидроксид -ионы, алкоксиды , амины и галогениды . Этот тип реакции встречается главным образом в алифатических углеводородах и редко в ароматических углеводородах . Последние имеют высокую электронную плотность и вступают в нуклеофильное ароматическое замещение только с очень сильными электроноакцепторными группами . Нуклеофильное замещение может происходить по двум различным механизмам: S N 1 и S N 2 . В их названиях S означает замещение, N — нуклеофильный, а число представляет кинетический порядок реакции: мономолекулярный или бимолекулярный. [45]

Реакция S N 1 протекает в две стадии. Сначала уходящая группа, удаляется образуя карбокатион . За этим следует быстрая реакция с нуклеофилом. [46]
В механизмах S N 2 нуклеофил образует переходное состояние с атакуемой молекулой, и только затем отщепляется уходящая группа. Эти два механизма различаются по стереохимии продуктов. S N 1 приводит к нестереоспецифическому присоединению и приводит не к хиральному центру, а скорее к набору геометрических изомеров ( цис/транс ). Напротив, обращение ( инверсия Вальдена 2 наблюдается в механизме S N ) существовавшей ранее стереохимии . [47]
Электрофильное замещение является аналогом нуклеофильного замещения, поскольку атакующий атом или молекула, электрофил , имеет низкую электронную плотность и, следовательно, положительный заряд. Типичными электрофилами являются атомы углерода карбонильных групп , карбокатионы или катионы серы или нитрония . Эта реакция протекает почти исключительно в ароматических углеводородах, где она называется электрофильным ароматическим замещением . Атака электрофилов приводит к образованию так называемого σ-комплекса — переходного состояния, в котором ароматическая система уничтожается. Затем уходящая группа, обычно протон, отщепляется и ароматичность восстанавливается. Альтернативой ароматическому замещению является электрофильное алифатическое замещение. Оно похоже на нуклеофильное алифатическое замещение и также имеет два основных типа: SE 1 и SE 2 . [48]

В третьем типе реакции замещения, радикальном замещении, атакующей частицей является радикал . [44] Этот процесс обычно принимает форму цепной реакции , например при реакции алканов с галогенами. На первом этапе свет или тепло разрушают галогенсодержащие молекулы с образованием радикалов. Далее реакция протекает лавинообразно, пока два радикала не встретятся и не рекомбинируются. [49]
- Реакции при цепной реакции радикального замещения


Добавление и исключение
Присоединение , представляют собой реакции , и его аналог, отщепление которые изменяют количество заместителей в атоме углерода и образуют или расщепляют кратные связи . Двойные и тройные связи могут быть получены путем исключения подходящей уходящей группы. Подобно нуклеофильному замещению, существует несколько возможных механизмов реакции, названных в честь соответствующего порядка реакции. В механизме E1 сначала выбрасывается уходящая группа, образуя карбокатион. Следующий этап — образование двойной связи — происходит с отщеплением протона ( депротонирование ). В механизме E1cb порядок ухода обратный, то есть первым отщепляется протон. Этот механизм требует участия базы. [50] Из-за схожести условий обе реакции элиминирования E1 или E1cb всегда конкурируют с замещением S N 1. [51]



Механизм Е2 также требует основания, но здесь атака основания и отщепление уходящей группы происходят одновременно и не приводят к образованию ионного промежуточного продукта. В отличие от элиминирования E1, для продукта реакции по механизму E2 возможны различные стереохимические конфигурации, поскольку атака основания происходит преимущественно в анти-положении по отношению к уходящей группе. Из-за сходства условий и реагентов элиминирование E2 всегда конкурирует с замещением S N 2 . [52]

Аналогом исключения является добавление, при котором двойные или тройные связи преобразуются в одинарные. Подобно реакциям замещения, существует несколько типов присоединения, различающихся типом атакующей частицы. Например, при электрофильном присоединении бромистого водорода электрофил (протон) атакует двойную связь, образуя карбокатион , который затем реагирует с нуклеофилом (бромом). Карбокатион может образовываться по обе стороны двойной связи в зависимости от групп, присоединенных к его концам, а предпочтительную конфигурацию можно предсказать с помощью правила Марковникова . [53] Это правило гласит: «При гетеролитическом присоединении полярной молекулы к алкену или алкину более электроотрицательный (нуклеофильный) атом (или часть) полярной молекулы присоединяется к атому углерода, несущему меньшее количество атомов водорода». [54]
Если присоединение функциональной группы происходит к менее замещенному атому углерода двойной связи, то электрофильное замещение кислотами невозможно. В этом случае приходится использовать реакцию гидроборирования-окисления , в которой на первом этапе атом бора действует как электрофил и присоединяется к менее замещенному атому углерода. На втором этапе нуклеофильный гидропероксид или анион галогена атакует атом бора. [55]
В то время как присоединение к богатым электронами алкенам и алкинам является в основном электрофильным, нуклеофильное присоединение играет важную роль в кратных связях углерод-гетероатом, и особенно в его наиболее важном представителе - карбонильной группе. Этот процесс часто связан с элиминированием, так что после реакции карбонильная группа снова присутствует. Поэтому она называется реакцией присоединения-отщепления и может происходить в производных карбоновых кислот, таких как хлориды, сложные эфиры или ангидриды. Эта реакция часто катализируется кислотами или основаниями, при этом кислоты увеличивают электрофильность карбонильной группы за счет связывания с атомом кислорода, тогда как основания усиливают нуклеофильность атакующего нуклеофила. [56]
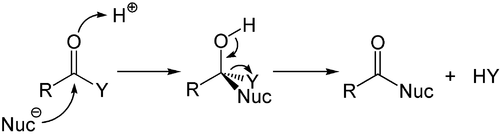
Нуклеофильное присоединение карбаниона , которая принадлежит к или другого нуклеофила к двойной связи альфа- , бета-ненасыщенного карбонильного соединения может происходить посредством реакции Михаэля более широкому классу конъюгатных присоединений . Это один из наиболее полезных методов мягкого образования связей C–C. [57] [58] [59]
Некоторые присоединения, которые невозможно осуществить с помощью нуклеофилов и электрофилов, можно осуществить с помощью свободных радикалов. Как и при свободнорадикальном замещении, радикальное присоединение протекает как цепная реакция, и такие реакции лежат в основе свободнорадикальной полимеризации . [60]
Другие механизмы органических реакций



В реакции перегруппировки углеродный скелет молекулы перестраивается с образованием структурного изомера исходной молекулы. К ним относятся реакции гидридного сдвига, такие как перегруппировка Вагнера-Меервейна , когда водородная , алкильная или арильная группа мигрирует от одного углерода к соседнему углероду. Большинство перегруппировок связано с разрывом и образованием новых углерод-углеродных связей. Другими примерами являются сигматропные реакции, такие как перегруппировка Коупа . [61]
Циклические перегруппировки включают циклоприсоединения и, в более общем смысле, перициклические реакции , в которых две или более молекулы, содержащие двойные связи, образуют циклическую молекулу. Важным примером реакции циклоприсоединения является реакция Дильса-Альдера (так называемое [4+2] циклоприсоединение) между сопряженным диеном и замещенным алкеном с образованием замещенной циклогексеновой системы. [62]
Будет ли происходить определенное циклоприсоединение, зависит от электронных орбиталей участвующих видов, поскольку только орбитали с одинаковым знаком волновой функции будут перекрываться и конструктивно взаимодействовать с образованием новых связей. Циклоприсоединению обычно способствует свет или тепло. Эти возмущения приводят к различному расположению электронов в возбужденном состоянии участвующих молекул и, следовательно, к различным эффектам. Например, реакциям [4+2] Дильса-Альдера может способствовать тепло, тогда как [2+2] циклоприсоединение избирательно индуцируется светом. [63] Из-за орбитального характера возможность образования стереоизомерных продуктов при циклоприсоединении ограничена, как описано правилами Вудворда-Гоффмана . [64]
Биохимические реакции
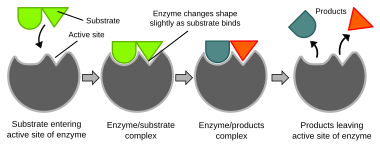
Биохимические реакции в основном контролируются сложными белками, называемыми ферментами , которые обычно специализируются на катализе только одной конкретной реакции. Реакция происходит в активном центре , небольшая часть фермента обычно находится в щели или кармане, выстланном аминокислотными остатками, а остальная часть фермента используется главным образом для стабилизации. Каталитическое действие ферментов зависит от нескольких механизмов, включая форму молекул («индуцированное соответствие»), напряжение связей, близость и ориентацию молекул относительно фермента, отдачу или отвод протонов (кислотно-основной катализ), электростатические взаимодействия и многие другие. [65]
Биохимические реакции, происходящие в живых организмах, называются метаболизмом . Среди наиболее важных его механизмов — анаболизм , при котором различные процессы, контролируемые ДНК и ферментами, приводят к производству крупных молекул, таких как белки и углеводы, из более мелких единиц. [66] Биоэнергетика изучает источники энергии для таких реакций. Важными источниками энергии являются глюкоза и кислород , которые могут вырабатываться растениями посредством фотосинтеза или усваиваться из пищи и воздуха соответственно. Все организмы используют эту энергию для производства аденозинтрифосфата (АТФ), который затем можно использовать для активизации других реакций. Разложение органического материала грибами , бактериями и другими микроорганизмами также входит в сферу биохимии .
Приложения

Химические реакции занимают центральное место в химической технологии , где они используются для синтеза новых соединений из природного сырья, такого как нефть , минеральные руды и кислород воздуха. Крайне важно сделать реакцию максимально эффективной, максимизируя выход и минимизируя количество реагентов, энергозатрат и отходов. Катализаторы особенно полезны для снижения энергии, необходимой для реакции, и увеличения скорости реакции . [67] [68]
Некоторые специфические реакции имеют свое нишевое применение. Например, термитная реакция используется для получения света и тепла в пиротехнике и сварке . Хотя она менее контролируема, чем более традиционная газокислородная сварка , дуговая сварка и оплавление , она требует гораздо меньше оборудования и до сих пор используется для ремонта рельсов, особенно в отдаленных районах. [69]
Мониторинг
Механизмы контроля химических реакций сильно зависят от скорости реакции. Относительно медленные процессы можно анализировать на месте на предмет концентраций и идентичности отдельных ингредиентов. Важными инструментами анализа в реальном времени являются измерение pH и анализ спектров оптического поглощения (цвета) и излучения. Менее доступный, но достаточно эффективный метод — введение в реакцию радиоактивного изотопа и наблюдение за тем, как он меняется с течением времени и куда перемещается; этот метод часто используют для анализа перераспределения веществ в организме человека. Более быстрые реакции обычно изучаются с помощью сверхбыстрой лазерной спектроскопии , где использование фемтосекундных лазеров позволяет отслеживать кратковременные переходные состояния за время, уменьшенное до нескольких фемтосекунд. [70]
См. также
- Химическое уравнение
- Химическая реакция
- Модель химической реакции
- Химик
- Химия
- Горение
- Ограничивающий реагент
- Список органических реакций
- Массовый баланс
- Микроскопическая обратимость
- Органическая реакция
- Кинетический анализ хода реакции
- Обратимая реакция
Отрасли химии |
|---|
Ссылки
- ^ ИЮПАК , Сборник химической терминологии , 2-е изд. («Золотая книга») (1997). Интернет-исправленная версия: (2006–) « Химическая реакция ». два : 10.1351/goldbook.C01033
- ^ Вейер, Дж. (1973). «Новые интерпретации алхимии». Химия в наше время . 7 (6): 177–181. дои : 10.1002/ciuz.19730070604 .
- ^ См. Ньюман, Уильям Р. (2004). Прометеевские амбиции: алхимия и поиски совершенной природы . Чикаго: Издательство Чикагского университета. ISBN 9780226575247 .
- ^ Краус, Пауль (1942–1943). Джабир ибн Хайян: Вклад в историю научных идей в исламе. I. Корпус джабирских сочинений. II. Джабир и греческая наука . Каир: Французский институт восточной археологии . ISBN 9783487091150 . OCLC 468740510 . , том. II, стр. 41–42.
- ^ Карпенко Владимир; Норрис, Джон А. (2002). «Купорос в истории химии» . Химические листы . 96 (12): 997–1005.
- ^ Фридман, Леонард Дж.; Фридман, Саманта Дж. (2008). История контактного сернокислотного процесса (PDF) . Бока-Ратон, Флорида: Acid Engineering & Consulting, Inc.
- ^ Стрэндж, Энтони Н. (2000). «Промышленность синтетического топлива Германии, 1935–1940». В Леше, Джон Э. (ред.). Немецкая химическая промышленность в двадцатом веке . Академическое издательство Kluwer . п. 170. ИСБН 978-0-7923-6487-0 .
- ^ Брок , стр. 34–55.
- ^ Брок , стр. 104–107.
- ^ Майерс, Ричард (2009). Основы химии . Издательская группа Гринвуд . п. 55. ИСБН 978-0-313-31664-7 .
- ^ ИЮПАК , Сборник химической терминологии , 2-е изд. («Золотая книга») (1997). Исправленная онлайн-версия: (2006–) « Уравнение химической реакции ». два : 10.1351/goldbook.C01034
- ^ Кори, Э.Дж. (1988). «Лекция Роберта Робинсона. Ретросинтетическое мышление? Основы и примеры». Обзоры химического общества . 17 : 111–133. дои : 10.1039/CS9881700111 .
- ^ ИЮПАК , Сборник химической терминологии , 2-е изд. («Золотая книга») (1997). Интернет-исправленная версия: (2006–) « Элементарная реакция ». дои : 10.1351/goldbook.E02035
- ^ Френкинг, Гернот (2006). «Элементарные реакции». Химический словарь Рёмппа . Тиме .
- ^ Jump up to: а б Кандори, Хидеки (2006). «Связывающие белки сетчатки». В Дугаве, Кристоф (ред.). Цис-транс-изомеризация в биохимии . Вайли-ВЧ . п. 56. ИСБН 978-3-527-31304-4 .
- ^ Аткинс , с. 114.
- ^ Аткинс , стр. 106–108.
- ^ Jump up to: а б "F*A*C*T - REACTION-Web" . www.crct.polymtl.ca .
- ^ Аткинс , с. 150
- ^ Аткинс , с. 963
- ^ Jump up to: а б с д Реагировать или не реагировать? Архивировано 10 января 2015 г. в Wayback Machine Управлении образования штата Юта . Проверено 4 июня 2011 г.
- ^ «8.3: Принцип Ле Шателье» . Химия LibreTexts . 05.08.2016 . Проверено 11 апреля 2023 г.
- ^ Jump up to: а б «11.5: Спонтанные реакции и свободная энергия» . Химия LibreTexts . 05.08.2016 . Проверено 11 апреля 2023 г.
- ^ «20.3: Спонтанные и неспонтанные реакции» . Химия LibreTexts . 27 июня 2016 г. Проверено 11 апреля 2023 г.
- ^ «Электролитические ячейки» . Химия LibreTexts . 02.10.2013 . Проверено 11 апреля 2023 г.
- ^ «Фотосинтез экзопланетных растений» . Химия LibreTexts . 26 мая 2016 г. Проверено 11 апреля 2023 г.
- ^ Уилбрахам, Энтони; Стэнли, Деннис; Уотерман, Эдвард; Матта, Майкл (2012). Химия . Пирсон. стр. 734–735. ISBN 9780132525763 .
- ^ Глускер, Дженни П. (1991). «Структурные аспекты лигандирования металлов с функциональными группами в белках». В Кристиане Б. Анфинсене (ред.). Достижения в области химии белков . Том. 42. Сан-Диего: Академик Пресс . п. 7. ISBN 978-0-12-034242-6 .
- ^ Го, Лян-Хун; Аллен, Х.; Хилл, О. (1991). «Прямая электрохимия белков и ферментов». В А.Г. Сайксе (ред.). Достижения неорганической химии . Том. 36. Сан-Диего: Академик Пресс . п. 359. ИСБН 978-0-12-023636-7 .
- ^ Виберг , стр. 289–290
- ^ Виберг , стр. 409.
- ^ Виберг , стр. 1180–1205 гг.
- ^ ИЮПАК , Сборник химической терминологии , 2-е изд. («Золотая книга») (1997). Исправленная онлайн-версия: (2006–) « Сопряженная кислотно-основная пара ». дои : 10.1351/goldbook.C01266
- ^ ИЮПАК , Сборник химической терминологии , 2-е изд. («Золотая книга») (1997). Интернет-исправленная версия: (2006–) « Осадки ». два : 10.1351/goldbook.P04795
- ^ Вингендер, Йорг; Ортандерл, Стефани (июль 2009 г.). «Осадки». Химический словарь Рёмппа . Тим .
- ^ Мейер, Х. Юрген (2007). «Химия твердого тела». В Эрвине Риделе (ред.). Современная неорганическая химия (на немецком языке) (3-е изд.). де Грюйтер . п. 171. ИСБН 978-3-11-019060-1 .
- ^ Винтерлин, Дж. (1997). «Атомные и макроскопические скорости реакций, катализируемых поверхностью». Наука . 278 (5345): 1931–4. Бибкод : 1997Sci...278.1931W . дои : 10.1126/science.278.5345.1931 . ПМИД 9395392 .
- ^ Вальдманн, Т.; Кюнцель, Д.; Хостер, HE; Гросс, А.; Бем, RJR (2012). «Окисление органического адслоя: вид с высоты птичьего полета». Журнал Американского химического общества . 134 (21): 8817–8822. дои : 10.1021/ja302593v . ПМИД 22571820 .
- ^ Аткинс , стр. 937–950.
- ^ Сондерс, Дэвид Стэнли (2002). Часы из насекомых (Третье изд.). Амстердам: Эльзевир . п. 179. ИСБН 978-0-444-50407-4 .
- ^ ИЮПАК , Сборник химической терминологии , 2-е изд. («Золотая книга») (1997). Интернет-исправленная версия: (2006–) « Катализатор ». doi : 10.1351/goldbook.C00876
- ^ ИЮПАК , Сборник химической терминологии , 2-е изд. («Золотая книга») (1997). Исправленная онлайн-версия: (2006–) « Ингибитор ». дои : 10.1351/goldbook.I03035
- ^ Эльшенбройх, Кристоф (2008). Металлоорганическая химия (6-е изд.). Висбаден: Vieweg+Teubner Verlag . п. 263. ИСБН 978-3-8351-0167-8 .
- ^ Jump up to: а б Марч, Джерри (1985), Продвинутая органическая химия: реакции, механизмы и структура, 3-е издание , Нью-Йорк: Wiley, ISBN 9780471854722 , OCLC 642506595
- ^ Хартсхорн, СР (1973). Алифатическое нуклеофильное замещение . Лондон: Издательство Кембриджского университета . п. 1. ISBN 978-0-521-09801-4 .
- ^ Бейтман, Лесли С.; Черч, Мервин Г.; Хьюз, Эдвард Д.; Ингольд, Кристофер К.; Тахер, Назир Ахмед (1940). «188. Механизм замещения у насыщенного атома углерода. Часть XXIII. Кинетическая демонстрация мономолекулярного сольволиза алкилгалогенидов. (Раздел Е) общее обсуждение». Журнал Химического общества : 979. doi : 10.1039/JR9400000979 .
- ^ Брюкнер , стр. 63–77.
- ^ Брюкнер , стр. 203–206.
- ^ Брюкнер , с. 16
- ^ Брюкнер , с. 192
- ^ Брюкнер , с. 183
- ^ Брюкнер , с. 172
- ^ Виберг , стр. 950, 1602
- ^ ИЮПАК , Сборник химической терминологии , 2-е изд. («Золотая книга») (1997). Исправленная онлайн-версия: (2006–) « Правило Марковникова ». дои : 10.1351/goldbook.M03707
- ^ Брюкнер , с. 125
- ^ Лача, Ганс Петер; Казмайер, Ули; Кляйн, Хельмут Альфонс (2008). Органическая химия: базовые знания по химии II (на немецком языке). Том 2 (6-е изд.). Спрингер . п. 273. ИСБН 978-3-540-77106-7 .
- ^ Дания, Скотт Э., изд. (2004). Органические реакции . дои : 10.1002/0471264180 . ISBN 978-0-471-26418-7 .
- ^ Хант, Ян. «Глава 18: Энолы и еноляты — реакция присоединения Михаэля» . Университет Калгари.
- ^ Брюкнер , с. 580
- ^ Лехнер, Манфред; Герке, Клаус; Нордмайер, Экхард (2003). Макромолекулярная химия (3-е изд.). Базель: Биркхойзер . стр. 53–65. ISBN 978-3-7643-6952-1 .
- ^ Фокс, Мэри Энн; Уайтселл, Джеймс К. (2004). Органическая химия (Третье изд.). Джонс и Бартлетт . п. 699. ИСБН 978-0-7637-2197-8 .
- ^ Дильс, О.; Олдер, К. (1928). «Синтезы гидроароматического ряда». «Анналы химии» Юстуса Либиха . 460 : 98–122. дои : 10.1002/jlac.19284600106 .
- ^ Брюкнер , стр. 637–647.
- ^ Вудворд, РБ; Хоффманн, Р. (1965). «Стереохимия электроциклических реакций». Журнал Американского химического общества . 87 (2): 395–397. дои : 10.1021/ja01080a054 .
- ^ Карлсон, Питер; Денеке, Детлеф; Колман, Ян; Фукс, Георг; Герок, Вольфганг (2005). Биохимия и патобиохимия Карлсона (на немецком языке) (16-е изд.). Тиме . стр. 55–56. ISBN 978-3-13-357815-8 .
- ^ ИЮПАК , Сборник химической терминологии , 2-е изд. («Золотая книга») (1997). Интернет-исправленная версия: (2006–) « Анаболизм ». два : 10.1351/goldbook.A00314
- ^ Эмиг, Герхард; Клемм, Элиас (2005). Техническая химия (на немецком языке) (5-е изд.). Спрингер . стр. 33–34. ISBN 978-3-540-23452-4 .
- ^ Трост, Б. (1991). «Атомная экономика – поиск синтетической эффективности». Наука . 254 (5037): 1471–1477. Бибкод : 1991Sci...254.1471T . дои : 10.1126/science.1962206 . ПМИД 1962206 .
- ^ Вейсмантель, Гай Э (1999). Джон Дж. МакКетта (ред.). Энциклопедия химической обработки и дизайна . Том. 67. ЦРК Пресс . п. 109. ИСБН 978-0-8247-2618-8 .
- ^ Аткинс , с. 987
Библиография
- Аткинс, Питер В.; Хулио де Паула (2006). Физическая химия (4-е изд.). Вайнхайм: Wiley-VCH . ISBN 978-3-527-31546-8 .
- Брок, Уильям Х. (1997). История химии Вьюга (на немецком языке). Брауншвейг: Vieweg . ISBN 978-3-540-67033-9 .
- Брюкнер, Рейнхард (2004). Механизмы реакции (на немецком языке) (3-е изд.). Мюнхен: Академическое издательство «Спектр». ISBN 978-3-8274-1579-0 .
- Виберг, Эгон, Виберг, Нильс и Холлеман, Арнольд Фредерик (2001). Неорганическая химия . Академическая пресса . ISBN 978-0-12-352651-9 .
{{cite book}}: CS1 maint: несколько имен: список авторов ( ссылка ) - . Британская энциклопедия . Том. 6 (11-е изд.). 1911. стр. 26–33.