Селегилин

Селегилин , также известный как L -депренил под торговыми марками Eldepryl , Zelapar и Emsam и продаваемый , среди прочего, , представляет собой лекарство , которое используется при лечении болезни Паркинсона и большого депрессивного расстройства . [4] [6] [8] [3] Он также был изучен по ряду других показаний, но не был официально одобрен для какого-либо другого использования. [20] [21] Лекарство в форме, разрешенной для лечения депрессии, имеет умеренную эффективность при этом состоянии, аналогичную эффективности других антидепрессантов . [21] [22] [23] Селегилин выпускается в виде проглатываемой таблетки или капсулы. [4] [5] или перорально распадающаяся таблетка (ODT) [6] [7] при болезни Паркинсона и в виде пластыря на кожу при депрессии. [8] [9]
Побочные эффекты селегилина, возникающие чаще, чем при приеме плацебо, включают бессонницу , среди прочего , , сухость во рту , головокружение , нервозность , ненормальные сновидения и реакции в месте применения (в форме пластыря). [21] [22] [24] [4] [8] В высоких дозах селегилин может вызывать опасные взаимодействия с пищей и лекарствами , такие как тирамин -связанная «сырная реакция» или гипертонический криз и риск серотонинового синдрома . [9] [25] [5] Однако дозы в пределах одобренного клинического диапазона, по-видимому, практически не имеют риска такого взаимодействия. [9][25][5] In addition, the ODT and transdermal patch forms of selegiline have reduced risks of such interactions compared to the conventional oral form.[7][9] Selegiline has no known misuse potential or dependence liability and is not a controlled substance.[26][27][28][29][8]
Selegiline acts as a monoamine oxidase inhibitor (MAOI) and thereby increases levels of monoamine neurotransmitters in the brain.[30][11][25][5] At typical clinical doses used for Parkinson's disease, selegiline is a selective and irreversible inhibitor of monoamine oxidase B (MAO-B), increasing brain levels of dopamine.[30][11][25][5] At higher doses, it loses its specificity for MAO-B and also inhibits monoamine oxidase A (MAO-A), which increases serotonin and norepinephrine levels in the brain as well.[30][11][25][5] In addition to its MAOI activity, selegiline is a catecholaminergic activity enhancer (CAE) and enhances the impulse-mediated release of norepinephrine and dopamine in the brain.[31][32][33][34][25] This action may be mediated by TAAR1 agonism.[35][36][37] After administration, selegiline partially metabolizes into levomethamphetamine and levoamphetamine, which act as norepinephrine releasing agents (NRAs) and may contribute to its therapeutic and adverse effects.[38][28][39] The levels of these metabolites are much lower with the ODT and transdermal patch forms of selegiline.[7][9] Chemically, selegiline is a substituted amphetamine,[40] a derivative of methamphetamine,[40] and the purified levorotatory enantiomer of deprenyl (the racemic form).[41][20]
Deprenyl was discovered and studied in the early 1960s.[41][20] Subsequently, selegiline was purified from deprenyl and was studied and developed itself.[41] Selegiline was first introduced for medical use in Hungary in 1977.[42] It was subsequently approved in the United Kingdom in 1982 and in the United States in 1989.[42][43] The ODT was approved in the United States in 2006 and in the European Union in 2010, while the patch was introduced in the United States in 2006.[42][20] Selegiline was the first selective MAO-B inhibitor to be discovered and marketed.[13][44][45] In addition to its medical use, there has been interest in selegiline as a potential anti-aging drug and nootropic.[46] However, effects of this sort are controversial and uncertain.[47][48][49][50] Generic versions of selegiline are available in the case of the conventional oral form but not in the case of the ODT or transdermal patch forms.[51][52]
Medical uses
[edit]Parkinson's disease
[edit]In its oral and ODT forms, selegiline is used to treat symptoms of Parkinson's disease (PD).[4][6] It is most often used as an adjunct to medications such as levodopa (L-DOPA), although it has been used off-label as a monotherapy.[53][54] The rationale for adding selegiline to levodopa is to decrease the required dose of levodopa and thus reduce the motor complications of levodopa therapy.[55] Selegiline delays the point when levodopa treatment becomes necessary from about 11 months to about 18 months after diagnosis.[56] There is some evidence that selegiline acts as a neuroprotective and reduces the rate of disease progression, though this is disputed.[54][55] In addition to parkinsonism, selegiline can improve symptoms of depression in people with Parkinson's disease.[57][58] There is evidence that selegiline may be more effective than rasagiline in the treatment of Parkinson's disease.[20][35][59] This may be due to pharmacological differences between the drugs, such as the catecholaminergic activity enhancer (CAE) actions of selegiline which rasagiline lacks.[20][35][59][32]
Depression
[edit]Selegiline is used as an antidepressant in the treatment of major depressive disorder (MDD).[8][21] Both the oral selegiline and transdermal selegiline patch formulations are used in the treatment of depression.[21] However, oral selegiline is not approved for depression and is used off-label for this indication, while the transdermal patch is specifically licensed for treatment of depression.[4][8] Both standard clinical doses of oral selegiline (up to 10 mg/day) and higher doses of oral selegiline (e.g., 30 to 60 mg/day) have been used to treat depression, with the lower doses selectively inhibiting MAO-B and the higher doses producing dual inhibition of both MAO-A and MAO-B.[9][21] Unlike oral selegiline, transdermal selegiline bypasses first-pass metabolism, thereby avoiding inhibition of gastrointestinal and hepatic MAO-A and minimizing the risk of food and drug interactions, whilst still allowing for selegiline to reach the brain and inhibit MAO-B.[9]
A 2023 systematic review and meta-analysis evaluated the effectiveness and safety of selegiline in the treatment of psychiatric disorders including depression.[21] It included both randomized and non-randomized published clinical studies.[21] The meta-analysis found that selegiline was more effective than placebo in terms of reduction in depressive symptoms (SMD = −0.96, k = 10, n = 1,308), response rates for depression improvement (RR = 1.61, k = 9, n = 1,238), and response rates for improvement of depression with atypical features (RR = 2.23, k = 3, n = 136).[21] Oral selegiline was significantly more effective than the selegiline patch in terms of depressive symptom improvement (SMD = −1.49, k = 6, n = 282 vs. SMD = −0.27, k = 4, n = 1,026, respectively; p = 0.03).[21] However, this was largely due to older and less methodologically rigorous trials that were at high risk for bias.[21] Oral selegiline studies also often employed much higher doses than usual, for instance 20 to 60 mg/day.[21] The quality of evidence of selegiline for depression was rated as very low overall, very low for oral selegiline, and low to moderate for transdermal selegiline.[21] For comparison, meta-analyses of other antidepressants for depression have found a mean effect size of about 0.3 (a small effect),[23][60] which is similar to that with transdermal selegiline.[21]
In two pivotal regulatory clinical trials of 6 to 8 weeks duration, the selegiline transdermal patch decreased scores on depression rating scales (specifically the 17- and 28-item HDRS) by 9.0 to 10.9 points, whereas placebo decreased scores by 6.5 to 8.6 points, giving placebo-subtracted differences attributable to selegiline of 2.4 to 2.5 points.[8] A 2013 quantitative review of the transdermal selegiline patch for depression, which pooled the results of these two trials, found that the placebo-subtracted number needed to treat (NNT) was 11 in terms of depression response (>50% reduction in symptoms) and 9 in terms of remission of depression (score of ≤10 on the MADRS).[22] For comparison, other antidepressants, including fluoxetine, paroxetine, duloxetine, vilazodone, adjunctive aripiprazole, olanzapine/fluoxetine, and extended-release quetiapine, have NNTs ranging from 6 to 8 in terms of depression response and 7 to 14 in terms of depression remission.[22] On the basis of these results, it was concluded that transdermal selegiline has similar effectiveness to other antidepressants.[22][61] NNTs are measures of effect size and indicate how many individuals would need to be treated in order to encounter one additional outcome of interest.[22] Lower NNTs are better, and NNTs corresponding to Cohen's d effect sizes have been defined as 2.3 for a large effect (d = 0.8), 3.6 for a medium effect (d = 0.5), and 8.9 for a small effect (d = 0.2).[22] The effectiveness of transdermal selegiline for depression relative to side effects and discontinuation was considered to be favorable.[22]
While several large regulatory clinical trials of transdermal selegiline versus placebo for depression have been conducted, there is a lack of trials comparing selegiline to other antidepressants.[52][61] Although multiple doses of transdermal selegiline were assessed, a dose–response relationship for depression was never established.[52][61] Transdermal selegiline has shown similar clinical effectiveness in the treatment of atypical depression relative to typical depression and in the treatment of anxious depression relative to non-anxious depression.[52][62][61]
Transdermal selegiline does not cause sexual dysfunction and may improve certain domains of sexual function, for instance sexual interest, maintaining interest during sex, and sexual satisfaction.[63] These benefits were apparent in women but not in men.[63] The lack of sexual dysfunction with transdermal selegiline is in contrast to many other antidepressants, such as the selective serotonin reuptake inhibitors (SSRIs) and serotonin–norepinephrine reuptake inhibitors (SNRIs), which are associated with high rates of sexual dysfunction.[64]
Transdermal selegiline patches have been underutilized in the treatment of depression compared to other antidepressants.[52][61] A variety of factors contributing to this underutilization have been identified.[52] One major factor is the very high cost of transdermal selegiline, which is often not covered by insurance and frequently proves to be prohibitive.[52][61] Conversely, other widely available antidepressants are much cheaper in comparison.[52][61]
Available forms
[edit]Selegiline is available in the following three pharmaceutical forms:[51]
- Oral tablets and capsules 5 mg (brand names Eldepryl, Jumex, and generics) – indicated for Parkinson's disease[4][5][42]
- Orally disintegrating tablets (ODTs) 1.25 mg (brand name Zelapar) – indicated for Parkinson's disease[6][7]
- Transdermal patches 6, 9, and 12 mg/24 hours (brand name Emsam) – indicated for major depressive disorder[8][9][12][24][61]

The transdermal patch form is also known as the "selegiline transdermal system" or "STS" and is applied once daily.[9][12][24][61][8] They are 20, 30, or 40 cm2 in size and contain a total of 20, 30, or 40 mg selegiline per patch (so 20 mg/20 cm2, 30 mg/30 cm2, and 40 mg/40 cm2), respectively.[8][61] The selegiline transdermal patch is a matrix-type adhesive patch with a three-layer structure.[8][61] It is the only approved non-oral MAOI, having reduced dietary restrictions and side effects in comparison to oral MAOIs, and is also the only approved non-oral first-line antidepressant.[61] The selegiline patch can be useful for those who have difficulty tolerating oral medications.[61]
Contraindications
[edit]Selegiline is contraindicated with serotonergic antidepressants including selective serotonin reuptake inhibitors (SSRIs), serotonin–norepinephrine reuptake inhibitors (SNRIs), and tricyclic antidepressants (TCAs), with serotonergic opioids like meperidine, tramadol, and methadone, with other monoamine oxidase inhibitors (MAOIs) such as linezolid, phenelzine, and tranylcypromine, and with dextromethorphan, St. John's wort, cyclobenzaprine, pentazocine, propoxyphene, and carbamazepine.[6][8][4] Combination of selegiline with serotonergic agents may cause serotonin syndrome, while combination of selegiline with adrenergic or sympathomimetic agents like ephedrine or amphetamines may cause hypertensive crisis.[6][8] Long washout periods are required before starting and stopping these medications with discontinuation or initiation of selegiline.[6][8][4][61]
Consumption of tyramine-rich foods can result in hypertensive crisis with selegiline, also known as the "cheese effect" or "cheese reaction" due to the high amounts of tyramine present in some cheeses.[6][11][44][65] Examples of other foods that may have high amounts of tyramine and similar substances include yeast products, chicken liver, snails, pickled herring, red wines, some beers, canned figs, broad beans, chocolate, and cream products.[65]
The preceding drug and food contraindications are dependent on selegiline dose and route, and hence are not necessarily absolute contraindications.[4][6][5][7][9] While high oral doses of selegiline (≥20 mg/day) can cause such interactions, oral doses within the approved clinical range (≤10 mg/day) appear to have little to no risk of these interactions.[9][25][5] In addition, the ODT and transdermal forms of selegiline have reduced risks of such interactions compared to the conventional oral form.[7][9]
Selegiline is also contraindicated in children less than 12 years of age and in people with pheochromocytoma, both due to heightened risk of hypertensive crisis.[8] For all human uses and all forms, selegiline is pregnancy category C, meaning that studies in pregnant animals have shown adverse effects on the fetus but there are no adequate studies in humans.[4][8]
Side effects
[edit]Side effects of the tablet form in conjunction with levodopa include, in decreasing order of frequency, nausea, hallucinations, confusion, depression, loss of balance, insomnia, increased involuntary movements, agitation, slow or irregular heart rate, delusions, hypertension, new or increased angina pectoris, and syncope.[4] Most of the side effects are due to a high dopamine levels, and can be alleviated by reducing the dose of levodopa.[3] Selegiline can also cause cardiovascular side effects such as orthostatic hypotension, hypertension, atrial fibrillation, and other types of cardiac arrhythmias.[66]
The main side effects of the patch form for depression include application-site reactions, insomnia, dry mouth, dizziness, nervousness, and abnormal dreams.[8][24] The selegiline patch carries a black box warning about a possible increased risk of suicide, especially for young people,[8] as do all antidepressants since 2007.[67]
Side effects of selegiline that have been identified as occurring significantly more often than with placebo in meta-analyses for psychiatric disorders have included dry mouth (RR = 1.58), insomnia (RR = 1.61, NNH = 19), and application site reactions with the transdermal form (RR = 1.81, NNH = 7).[21][22] No significant diarrhea, headache, dizziness, nausea, sexual dysfunction, or weight gain were apparent in these meta-analyses.[21][22]
Selegiline, including in its oral, ODT, and patch forms, has been found to cause hypotension or orthostatic hypotension in some individuals.[4][6][8] In a clinical trial, the rate of systolic orthostatic hypotension was 21% versus 9% with placebo and the rate of diastolic orthostatic hypotension was 12% versus 4% with placebo in people with Parkinson's disease taking the ODT form of selegiline.[6] The risk of hypotension is greater at the start of treatment and in the elderly (3% vs. 0% with placebo).[6] The rate of hypotension or orthostatic hypotension with the selegiline patch was 2.2% versus 0.5% with placebo in clinical trials of people with depression.[24] Significant orthostatic blood pressure changes (≥10 mm Hg decrease) occurred in 9.8% versus 6.7% with placebo, but most of these cases were asymptomatic and heart rate was unchanged.[24][68] The rates of other orthostatic hypotension-related side effects in this population were dizziness or vertigo 4.9% versus 3.1% with placebo and fainting 0.5% versus 0.0% with placebo.[24] It is said that orthostatic hypotension is rarely seen with the selegiline transdermal patch compared to oral MAOIs.[52] Caution is advised against rapidly rising after sitting or lying, especially after prolonged periods or at the start of treatment, as this can result in fainting.[6][27][68] Falls are of particular concern in the elderly.[68] MAOIs like selegiline may lower blood pressure by increasing dopamine levels and activating dopamine receptors, by increasing levels of the false neurotransmitter octopamine, and/or by other mechanisms.[69]
Meta-analyses published in the 1990s found that the addition of selegiline to levodopa increased mortality in people with Parkinson's disease.[27] However, several subsequent meta-analyses with more trials and patients found no increase in mortality with selegiline added to levodopa.[27][70][71] If selegiline does increase mortality, it has been theorized that this may be due to cardiovascular side effects, such as its amphetamine-related sympathomimetic effects and its MAO inhibition-related hypotension.[72] Although selegiline does not seem to increase mortality, it appears to worsen cognition in people with Parkinson's disease over time.[73] Conversely, rasagiline does not seem to do so and can enhance cognition.[73]
Rarely, selegiline has been reported to induce or exacerbate impulse control disorders, pathological gambling, hypersexuality, and paraphilias in people with Parkinson's disease.[74][75][76][77][78][79][80][81] However, MAO-B inhibitors like selegiline causing impulse control disorders is uncommon and controversial.[74][75] Selegiline has also been reported to activate or worsen rapid eye movement (REM) sleep behavior disorder (RBD) in some people with Parkinson's disease.[82][83][84]
Selegiline has shown little or no misuse potential in humans or monkeys.[26][27][28][85][86][87] Likewise, it has no dependence potential in rodents.[29] This is in spite of its amphetamine active metabolites, levomethamphetamine and levoamphetamine, and is in contrast to agents like dextroamphetamine and dextromethamphetamine.[27][28][29][86][87] However, selegiline can strongly potentiate the reinforcing effects of exogenous β-phenethylamine by inhibiting its MAO-B-mediated metabolism.[28] Misuse of the combination of selegiline and β-phenethylamine has been reported.[88][89]
Overdose
[edit]Little information is available about clinically significant selegiline overdose.[4] The drug has been studied clinically at doses as high as 60 mg/day orally,[90][21] 10 mg/day as an ODT,[7] and 12 mg/24 hours as a transdermal patch.[9] In addition, deprenyl (the racemic form) has been clinically studied orally at doses as large as 100 mg/day.[30] During clinical development of oral selegiline, some individuals who were exposed to doses of 600 mg developed severe hypotension and psychomotor agitation.[4][6] Overdose may result in non-selective inhibition of both MAO-A and MAO-B and may be similar to overdose of other non-selective monoamine oxidase inhibitors (MAOIs) like phenelzine, isocarboxazid, and tranylcypromine.[4][6] Serotonin syndrome, hypertensive crisis, and/or death may occur with overdose.[4][6][8] No specific antidote to selegiline overdose is available.[8]
Interactions
[edit]Serotonin syndrome and hypertensive crisis
[edit]Both the oral and patch forms of selegiline come with strong warnings against combining it with drugs that could produce serotonin syndrome, such as selective serotonin reuptake inhibitors (SSRIs) and the cough medicine dextromethorphan.[4][8][91] Selegiline in combination with the opioid analgesic pethidine is not recommended, as it can lead to severe adverse effects.[91] Several other synthetic opioids such as tramadol and methadone, as well as various triptans, are also contraindicated due to potential for serotonin syndrome.[92][93]
All three forms of selegiline carry warnings about food restrictions to avoid hypertensive crisis that are associated with MAOIs.[4][6][8] The patch form was created in part to overcome food restrictions; clinical trials showed that it was successful.[22][8] Additionally, in post-marketing surveillance from April 2006 to October 2010, only 13 self-reports of possible hypertensive events or hypertension were made out of 29,141 exposures to the drug, and none were accompanied by objective clinical data.[22] The lowest dose of the patch method of delivery, 6 mg/24 hours, does not require any dietary restrictions.[94] Higher doses of the patch and oral formulations, whether in combination with the older non-selective MAOIs or in combination with the reversible MAO-A inhibitor (RIMA) moclobemide, require a low-tyramine diet.[91]
A study found that selegiline in transdermal patch form did not importantly modify the pharmacodynamic effects or pharmacokinetics of the sympathomimetic agents pseudoephedrine and phenylpropanolamine.[9][95] Likewise, oral selegiline at an MAO-B-selective dosage did not appear to modify the pharmacodynamic effects or pharmacokinetics of intravenous methamphetamine in another study.[96][97] Conversely, selegiline, also at MAO-B-selective doses, has been found to reduce the physiological and euphoric subjective effects of cocaine whilst not affecting its pharmacokinetics in some studies but not in others.[98][99][100][101][102][103] Cautious safe combination of MAOIs like selegiline with stimulants like lisdexamfetamine has been reported.[104][105][106] However, a hypertensive crisis with selegiline and ephedrine has also been reported.[4] The selegiline drug labels warn about combination of selegiline with indirectly-acting sympathomimetic agents, like amphetamines, ephedrine, pseudoephedrine, and phenylpropanolamine, due to the potential risk of hypertensive crisis, and recommend monitoring blood pressure with such combinations.[6][8] The combination of selegiline with certain other medications, like phenylephrine and buspirone, is also warned against for similar reasons.[8][12][107][68] In the case of phenylephrine, this drug is substantially metabolized by monoamine oxidase, including by both MAO-A and MAO-B.[108][109]
Besides norepinephrine releasing agents, selective norepinephrine reuptake inhibitors (NRIs) may be safe in combination with MAOIs like selegiline.[110][111][112] Potent NRIs, such as reboxetine, desipramine, protriptyline, and nortriptyline, can reduce or block the pressor effects of tyramine, including in those taking MAOIs.[110][111][112] This is by inhibiting the norepinephrine transporter (NET) and preventing entry of tyramine into presynaptic noradrenergic neurons where tyramine induces the release of norepinephrine.[110][111][112] As a result, NRIs may reduce the risk of tyramine-related hypertensive crisis in people taking MAOIs.[110][111][112] Norepinephrine–dopamine reuptake inhibitors (NDRIs), like methylphenidate and bupropion, are also considered to be safe in combination with MAOIs.[113] However, initiation at low doses and slow upward dose titration is advisable in the case of both NRIs and NDRIs due to possible potentiation of their effects and side effects by MAOIs.[113]
Cytochrome P450 inhibitors and inducers
[edit]The cytochrome P450 enzymes involved in the metabolism of selegiline have not been fully elucidated.[5][18] CYP2D6 and CYP2C19 metabolizer phenotypes did not significantly affect the pharmacokinetics of selegiline, suggesting that these enzymes are minimally involved in its metabolism and that inhibitors and inducers of these enzymes would not importantly affect its pharmacokinetics.[18][40][114][115] However, although most pharmacokinetic variables were unaffected, overall exposure to selegiline's metabolite levomethamphetamine was 46% higher in CYP2D6 poor metabolizers compared to extensive metabolizers and exposure to its metabolite desmethylselegiline was 68% higher in CYP2C19 poor metabolizers compared to extensive metabolizers.[40][114][115] As with the cases of CYP2D6 and CYP2C19, the strong CYP3A4 and CYP3A5 inhibitor itraconazole has minimal impact on the pharmacokinetics of selegiline, suggesting lack of major involvement of this enzyme as well.[18][116][6] On the other hand, the anticonvulsant carbamazepine, which is known to act as a strong inducer of CYP3A enzymes,[117] has paradoxically been found to increase exposure to selegiline and its metabolites levomethamphetamine and levoamphetamine by approximately 2-fold (with selegiline used as the transdermal patch form).[8][9] One enzyme thought to be majorly involved in the metabolism of selegiline based on in-vitro studies is CYP2B6.[5][18][9][19] However, there are no clinical studies of different CYP2B6 metabolizer phenotypes or of CYP2B6 inhibitors or inducers on the pharmacokinetics of selegiline.[44] In addition to CYP2B6, CYP2A6 may be involved in the metabolism of selegiline to a lesser extent.[44][118]
Birth control pills containing the synthetic estrogen ethinylestradiol and a progestin like gestodene or levonorgestrel have been found to increase peak levels and overall exposure to oral selegiline by 10- to 20-fold.[18][119][120] High levels of selegiline can lead to loss of MAO-B selectivity and inhibition of MAO-A as well.[18][120] This increases susceptibility to side effects and interactions of non-selective monoamine oxidase inhibitors (MAOIs), such as tyramine-induced hypertensive crisis and serotonin toxicity when combined with serotonergic medications.[18][120] However, this study had a small sample size of four individuals as well as other methodological limitations.[18][120] The precise mechanism underlying the interaction is unknown, but is likely related to cytochrome P450 inhibition and consequent inhibition of selegiline first-pass metabolism by ethinylestradiol.[18] In contrast to birth control pills containing ethinylestradiol, menopausal hormone therapy with estradiol and levonorgestrel did not modify peak levels of selegiline and only modestly increased overall exposure (+59%).[18][119][121] Hence, menopausal hormone therapy does not pose the same risk of interaction as ethinylestradiol-containing birth control pills when taken together with selegiline.[119][121]
Overall exposure to selegiline with oral selegiline has been found to be 23-fold lower in people taking anticonvulsants known to strongly activate drug-metabolizing enzymes.[122] The anticonvulsants included phenobarbital, phenytoin, carbamazepine, and amobarbital.[122] In a previous study however, carbamazepine specifically did not reduce selegiline exposure.[8][9] Phenobarbital and certain other anticonvulsants are known to strongly induce CYP2B6, one of the major enzymes believed to be involved in selegiline metabolism.[122] As such, it was concluded that strong CYP2B6 induction was most likely responsible for the dramatically reduced exposure to selegiline observed in the study.[122]
Selegiline inhibition of cytochrome P450 enzymes
[edit]Selegiline has been reported to inhibit several cytochrome P450 enzymes, including CYP2D6, CYP3A4/5, CYP2C19, CYP2B6, and CYP2A6.[8][123] It is a mechanism-based inhibitor (suicide inhibitor) of CYP2B6 and has been said to "potently" or "strongly" inhibit this enzyme in vitro.[124][123][125][126] It may inhibit the metabolism of bupropion, a major CYP2B6 substrate, into its active metabolite hydroxybupropion.[124][123][125] However, a study predicted that inhibition of CYP2B6 by selegiline would non-significantly affect exposure to bupropion.[126] Selegiline has not been listed or described as a clinically significant CYP2B6 inhibitor by the Food and Drug Administration (FDA) as of 2023.[117][8] One small study observing three patients found that selegiline was safe and well-tolerated in combination with bupropion.[125][127] In addition to CYP2B6 and other cytochrome P450 enzymes, selegiline is a potent mechanism-based inhibitor of CYP2A6 and may increase exposure to nicotine (a major CYP2A6 substrate).[128][129] By inhibiting cytochrome P450 enzymes like CYP2B6 and CYP1A2, selegiline may inhibit its own metabolism and thereby interact with itself.[129][130]
Other interactions
[edit]Dopamine antagonists like antipsychotics or metoclopramide, which block dopamine receptors and thereby antagonize the dopaminergic effects of selegiline, could potentially reduce the effectiveness of the medication.[6] Dopamine-depleting agents like reserpine and tetrabenazine, by reducing dopamine levels, can also oppose the effectiveness of dopaminergic medications like selegiline.[131]
Pharmacology
[edit]Pharmacodynamics
[edit]Monoamine oxidase inhibitor
[edit]Selegiline acts as an enzyme inhibitor of the enzyme monoamine oxidase (MAO) and hence is known as a monoamine oxidase inhibitor (MAOI).[30][11][25][5] There are two types of MAO, MAO-A and MAO-B.[30][11][25][5] MAO-A metabolizes the monoamine neurotransmitters serotonin, dopamine, and norepinephrine as well as trace amines like tyramine, whereas MAO-B metabolizes dopamine and the trace amine β-phenethylamine.[30][11][25][5] At lower concentrations and at typical clinical doses (≤10 mg/day), selegiline selectively inhibits MAO-B.[30][11][25][5] Conversely, at higher concentrations and doses (≥20 mg/day), selegiline additionally inhibits MAO-A.[30][11][25][5] By selectively inhibiting MAO-B, selegiline increases levels of dopamine in the brain and thereby increases dopaminergic neurotransmission.[30][11][25][5] At higher doses, by inhibiting both MAO-A and MAO-B, selegiline increases brain levels of serotonin, dopamine, and norepinephrine and thereby increases serotonergic, dopaminergic, and noradrenergic neurotransmission.[30][11][25][5] Selegiline is an irreversible mechanism-based inhibitor (suicide inhibitor) of MAO that acts by covalently binding to the active site of the enzyme and thereby disabling it.[30][11][25][5][66]
Selegiline is thought to exert its therapeutic effects in the treatment of the motor symptoms of Parkinson's disease by increasing dopamine levels in the substantia nigra pars compacta (SNpc) of the basal ganglia, which projects to the caudate nucleus and putamen of the striatum, thereby enhancing the signaling of the nigrostriatal pathway.[66][30][132][133][17] In addition to the nigrostriatal pathway, selegiline may also influence and potentiate other dopaminergic pathways and areas, including the mesolimbic pathway, mesocortical pathway, tuberoinfundibular pathway, and chemoreceptor trigger zone, which may also be involved in its effects as well as side effects.[134][135][136] Selegiline and other MAO-B inhibitors may additionally improve non-motor symptoms in Parkinson's disease, for instance depression and motivational deficits, by increasing dopamine levels.[66] Selegiline may have some disease-modifying neuroprotective effects in Parkinson's disease by inhibiting the MAO-B-mediated oxidation of dopamine into reactive oxygen species that damage dopaminergic neurons in the nigrostriatal pathway via oxidative stress.[137][66] However, the pathophysiology of Parkinson's disease is complex and multifacted, and MAO-B inhibitors may only slow the progression of the disease and do not halt it.[137][66]
Selegiline almost completely inhibits MAO-B in blood platelets at a dosage of 10 mg/day.[7] Following a single 5 or 10 mg oral dose of selegiline, 86 to 90% of MAO-B activity in platelets was inhibited within 2 to 4 hours and 98% of activity was inhibited after 24 hours.[5][30] Inhibition of platelet MAO-B activity persisted at above 90% for 5 days and almost 14 days were required before activity returned to baseline.[5][30] A lower dose of selegiline of 1 mg/day for 10 days also inhibited platelet MAO-B activity by about 75 to 100% in three individuals.[30][138] Similarly, 2.5 mg/day selegiline inhibited platelet MAO-B by 95% within 4 days.[139] The recommended dosing schedule of selegiline in Parkinson's disease (10 mg/day) has been described as somewhat questionable and potentially excessive from a pharmacological standpoint.[140][139] Selegiline could be effective at lower doses, like 2.5 mg/day.[141][139] However, optimal effectiveness of selegiline in Parkinson's disease seems to require a dosage of 10 mg/day and its effectiveness lasts only about 2 to 3 days following discontinuation.[30][142] It is assumed that peripheral and brain MAO-B are inhibited with selegiline to similar extents.[50][69][30] Accordingly, selegiline at an MAO-B-selective dosage of 10 mg/day has been found to inhibit brain MAO-B by more than 90% in postmortem individuals with Parkinson's disease.[13][38][143][144] This dosage of selegiline has been found in such individuals to produce increases in brain levels of dopamine of 23 to 350% and of β-phenethylamine of 1,200 to 3,400% depending on the brain area and the study.[25][30][145][143][146][147] Brain MAO-B levels recover slowly upon discontinuation of selegiline, with a half-time of brain MAO-B synthesis and recovery of approximately 40 days in humans.[25][144]
Selegiline is about 500 to 1,000 times more potent in inhibiting MAO-B than MAO-A in vitro and about 100 times more potent in vivo in rodents.[30][11][50] The clinical selectivity of selegiline for MAO-B is lost at doses of the drug above 20 mg/day.[30] In a study of post-mortem individuals who were on selegiline 10 mg/day, MAO-A activity in the brain was inhibited by 38 to 86%.[30][25] A more recent study using positron emission tomography (PET) imaging similarly found inhibition of brain MAO-A by 33 to 70% in humans.[42][148] However, while brain dopamine and β-phenethylamine levels are substantially increased at this dosage, brain levels of serotonin and its metabolite 5-hydroxyindoleacetic acid (5-HIAA) remain unchanged.[25][30][145] It has been found in animal studies that brain MAO-A must be inhibited by nearly 85% before serotonin, norepinephrine, or dopamine levels increase and result in increased functional activity as well as accompanying behavioral changes.[25][149] Selegiline at an oral dosage of 10 mg/day does not cause the "cheese effect" as assessed by oral tyramine and β-phenethylamine challenge tests.[5] These findings indicate that selegiline does not importantly inhibit MAO-A at a dosage of 10 mg/day.[5] However, a dosage of 20 mg/day selegiline did increase the pressor effect of tyramine, indicating that doses this high and above can significantly inhibit MAO-A.[25] The "cheese reaction" is known to be specifically dependent on inhibition of intestinal MAO-A.[30][25]
Besides increasing brain dopamine levels via MAO-B inhibition, selegiline strongly increases endogenous levels of β-phenethylamine, a major substrate of MAO-B.[30] Levels of β-phenethylamine in the brain are increased 10- to 30-fold and levels in urine are increased 20- to 90-fold.[30][145][150] β-Phenethylamine is normally present in small amounts in the brain and urine and has been referred to as "endogenous amphetamine".[30][151] Similarly to amphetamines, it induces the release of norepinephrine and dopamine and produces psychostimulant effects.[30] Selegiline also strongly increases levels of β-phenethylamine with exogenous administration of β-phenethylamine.[30] The increase in endogenous levels of β-phenethylamine with selegiline might be involved in its effects, for instance claimed "psychic energizing" and mood-lifting effects as well as its effectiveness in the treatment of Parkinson's disease.[152][46][153] In contrast to amphetamine psychostimulants however, selegiline is thought to have little or no misuse potential.[46][85]
The MAO-B inhibition of deprenyl lies mainly in selegiline (L-deprenyl), which is 150-fold more potent than D-deprenyl at inhibiting MAO-B.[38][154] Besides selegiline itself, desmethylselegiline, one of its major metabolites, is pharmacologically active.[38][155] Compared to selegiline, desmethylselegiline is 60-fold less potent in inhibiting MAO-B in vitro, but is only 3- to 6-fold less potent in vivo.[5][155] Although desmethylselegiline levels with selegiline therapy are low, selegiline and desmethylselegiline are highly potent MAO-B inhibitors due to the irreversible nature of their inhibition.[30] As such, desmethylselegiline may contribute significantly to the MAO-B inhibition with selegiline.[30]
Findings from a 2021 study suggest that MAO-A is solely or almost entirely responsible for the striatal metabolism of dopamine rather than MAO-B.[156][157][158] Conversely, MAO-B was found to regulate tonic γ-aminobutyric acid (GABA) levels.[156][157][158] These findings may warrant a rethinking of the pharmacological actions of MAO-B inhibitors like selegiline in the treatment of Parkinson's disease.[156][157][158]
Catecholaminergic activity enhancer
[edit]Selegiline has been found to act as a catecholaminergic activity enhancer (CAE).[31][32][33] It selectively enhances the activity of noradrenergic and dopaminergic neurons and does not affect the activity of serotonergic neurons.[159][34][25] The CAE actions of selegiline are distinct from those of catecholamine releasing agents like amphetamines.[31][32][33] Conversely, the actions are shared with certain trace amines like β-phenethylamine and tryptamine.[160][36] Selegiline and other CAEs enhance only impulse propogation-mediated release of catecholamines.[31][33] In relation to this, they lack the misuse potential of amphetamines.[31][32] Selegiline is active as a CAE at far lower concentrations and doses than those at which it starts to inhibit the monoamine oxidases.[159][161][25] For example, selegiline given subcutaneously in rodents selectively inhibits MAO-B with a single dose of at least 0.2 mg/kg, whereas CAE effects are apparent for noradrenergic neurons at a dose of 0.01 mg/kg (+42% activity) and for dopaminergic neurons at a dose of 0.025 mg/kg (+17% activity) (i.e., 8- to 20-fold lower doses).[25][note 1][159] Monoamine activity enhancers (MAEs) show a peculiar and characteristic bimodal concentration–response relationship, with two bell-shaped curves of activity across tested concentration ranges.[36][163][159][164] Selegiline is presently the only registered pharmaceutical medication with CAE actions that lacks concomitant potent catecholamine releasing effects.[160][159][165]
Other MAEs besides selegiline, like phenylpropylaminopentane (PPAP) and benzofuranylpropylaminopentane (BPAP), have been developed.[33][160] PPAP was derived from selegiline (and by extension from β-phenethylamine), while BPAP was derived from tryptamine.[160] These compounds are more potent and selective in their MAE actions than selegiline.[160][36] In addition, BPAP is an activity enhancer of not only catecholaminergic neurons but also of serotonergic neurons.[34] Unlike selegiline, PPAP and BPAP lack the MAO inhibition and amphetamine metabolites of selegiline, although BPAP has also been found to inhibit the reuptake of dopamine, norepinephrine, and serotonin.[160][166]
The actions of MAEs including selegiline may be due to TAAR1 agonism.[167][35] TAAR1 agonists have been found to enhance the release of monoamine neurotransmitters like dopamine and serotonin analogously to MAEs;[168][169][35] trace amines like β-phenethylamine and tryptamine are known to act as both TAAR1 agonists and MAEs;[168][169] and the TAAR1 antagonist EPPTB has been shown to reverse the CAE effects of BPAP and selegiline, among other findings.[167][35] However, it has yet to be determined whether MAEs like BPAP and selegiline actually directly bind to and activate the TAAR1.[37][35] Moreover, in an older study of MAO-B knockout mice, no non-MAO binding of radiolabeled selegiline was detected in the brain, suggesting that this agent might not act directly via a macromolecular target in terms of its MAE effects.[170][171][172] In any case, selegiline's active metabolites levomethamphetamine and levoamphetamine have been confirmed to bind to and activate the TAAR1.[173][174][175] As with selegiline, levomethamphetamine and levoamphetamine are also CAEs, although levomethamphetamine is 1- to 10-fold less potent in this action than selegiline itself.[32][25][176][177][160][35] Another metabolite of selegiline, desmethylselegiline, has been found to act as a CAE as well.[178][179] TAAR1 agonists like ulotaront and ralmitaront are under investigation for treatment of a variety of psychiatric disorders, such as depression and schizophrenia.[180][181]
In contrast to selegiline, rasagiline is devoid of CAE actions.[20][178] In fact, it actually inhibits the CAE effects of selegiline.[35] This may explain differences in effectiveness between selegiline and rasagiline in the treatment of Parkinson's disease.[20][35][59] According to József Knoll, one of the original developers of selegiline, the CAE effect of selegiline may be more important than MAO-B inhibition in terms of effectiveness for Parkinson's disease.[32] Rasagiline may act as a TAAR1 antagonist to mediate its anti-CAE effects.[159][35] However, as with selegiline, binding to and modulation of the TAAR1 by rasagiline still requires confirmation.[35]
Selegiline has potent pro-sexual or aphrodisiac effects in male rodents.[30][182][183][184] The pro-sexual effects of selegiline are stronger than those of dopamine agonists like apomorphine and bromocriptine and high doses of amphetamine.[30][182][184] These effects are not shared with other MAO-B inhibitors or the MAO-A inhibitor clorgiline and hence do not appear to be related to MAO inhibition.[30][183] Instead, the CAE actions of selegiline have been implicated in the pro-sexual effects.[20][160] Although selegiline has shown potent pro-sexual effects in rodents, these effects were not subsequently confirmed in primates.[30][185] In humans, selegiline for depression shows minimal pro-sexual effects in men, though it did significantly enhance several areas of sexual function in women.[63] However, this may have been due to improvement in depression.[63]
Catecholamine releasing agent
[edit]Levomethamphetamine and levoamphetamine are major metabolites of selegiline and are also pharmacologically active.[38][39] They are sympathomimetic and psychostimulant agents that work by inducing the release of norepinephrine and dopamine in the body and brain.[38][39][186]
The involvement of levomethamphetamine and levoamphetamine in the effects of selegiline is controversial.[28] The levels of these metabolites are relatively low and are potentially below pharmacological concentrations at typical clinical doses of selegiline.[30][4] In any case, both beneficial and harmful effects of these metabolites have been postulated.[28] It is unknown whether the metabolites are involved in the effectiveness of selegiline in the treatment of Parkinson's disease.[30] It has been said that the amphetamine metabolites of selegiline might improve fatigue, but could also produce cardiovascular side effects like increased heart rate and blood pressure and reportedly may be able to cause insomnia, euphoria, psychiatric disturbances, and psychosis.[38][7][17] It is unknown what concentrations of levomethamphetamine and levoamphetamine produce sympathomimetic and other effects in humans and whether such concentrations are achieved with selegiline therapy.[38] However, cardiovascular side effects of selegiline have been found clinically and have been attributed to its amphetamine metabolites.[187][50] For comparison, rasagiline, which lacks amphetamine metabolites, has shown fewer adverse effects in clinical studies.[187][50][188] Animal studies suggest that selegiline's amphetamine metabolites may indeed be involved in its effects, such as arousal, wakefulness, locomotor activity, and sympathomimetic effects.[189][190][153][191][192]
Whereas the psychostimulants dextromethamphetamine and dextroamphetamine are relatively balanced releasers of dopamine and norepinephrine, levomethamphetamine is about 15- to 20-fold more potent in releasing norepinephrine relative to dopamine in vitro.[193][39][194][195][196] Levomethamphetamine and levoamphetamine are similar to dextromethamphetamine and dextroamphetamine in their potencies as norepinephrine releasers in rodents in vivo.[186][197][196][198] Conversely, levomethamphetamine is dramatically less potent as a dopamine releaser than dextromethamphetamine in vivo, whereas levoamphetamine is 3- to 5-fold less potent as a dopamine releaser compared to dextroamphetamine.[197][186][198] Relatedly, levoamphetamine is substantially more potent as a dopamine releaser and stimulant than levomethamphetamine in rodents.[197][198] In relation to the preceding findings, levomethamphetamine acts more as a selective norepinephrine releasing agent and levoamphetamine as an imbalanced and norepinephrine-preferring releasing agent of norepinephrine and dopamine than as balanced dual releasers of these catecholamine neurotransmitters.[39][197][186][195][38][194] In accordance with the results of catecholamine release studies, levomethamphetamine is 2- to 10-fold or more less potent than dextromethamphetamine in terms of psychostimulant-like effects in rodents,[199][200][201] whereas levoamphetamine is 1- to 4-fold less potent than dextroamphetamine in its stimulating and reinforcing effects in monkeys and humans.[186][30][202]
In clinical studies, levomethamphetamine at oral doses of 1 to 10 mg has been found not to affect subjective drug responses, heart rate, blood pressure, core temperature, electrocardiography, respiration rate, oxygen saturation, or other clinical parameters.[203][204] As such, doses of levomethamphetamine of less than or equal to 10 mg appear to have no significant physiological or subjective effects.[203][204] However, higher doses of levomethamphetamine, for instance 0.25 to 0.5 mg/kg (mean doses of ~18–37 mg) intravenously, have been reported to produce significant pharmacological effects, including increased heart rate and blood pressure, increased respiration rate, and subjective effects like intoxication and drug liking.[203][195] On the other hand, in contrast to dextroamphetamine and dextromethamphetamine, levomethamphetamine also produces subjective "bad" or aversive drug effects.[194][195] Unlike the case of levomethamphetamine, oral doses of levoamphetamine of as low as 5 mg and above have been assessed and reported to produce significant pharmacological effects, for instance on wakefulness and mood.[205][202][note 2][206][207] With a 10 mg oral dose of selegiline, about 2 to 6 mg levomethamphetamine and 1 to 3 mg levoamphetamine is excreted in urine.[7][5][17]
The amphetamine metabolites of selegiline being involved in its effectiveness in the treatment of Parkinson's disease has been deemed unlikely.[30] High doses of levoamphetamine, for instance 50 mg/day, have been reported to be slightly effective in the treatment of Parkinson's disease.[30][17][207] It has been postulated that amphetamines are limitedly effective for Parkinson's disease as there is inadequate presynaptic dopamine to be released in patients with the condition.[205][207] In any case, this effectiveness of high doses of levoamphetamine could not be relevant to selegiline, which is administered at a dose of 10 mg/day.[30] In one clinical study, levels of the amphetamine metabolites of selegiline were manipulated and there were no changes in clinical symptoms of Parkinson's disease.[30][208] This led the researchers to conclude that the beneficial clinical effects of selegiline in Parkinson's disease were not due to its amphetamine metabolites.[30][208] It is possible that there could be some small synergistic beneficial effect of selegiline with its amphetamine metabolites, but this has been considered improbable.[30]
Methamphetamine is directly neurotoxic to dopaminergic neurons at high concentrations and doses.[209] Such toxicity is unfavorable generally, but it is particularly concerning in the context of Parkinson's disease due to the potential for sufficiently high concentrations of methamphetamine to further exarcebate neurodegeneration along the nigrostriatal pathway.[210][211][28] However, as previously described, levomethamphetamine is a significantly weaker monoamine releaser and psychostimulant than dextromethamphetamine.[210][39][28] Circulating levels of levomethamphetamine associated with clinically relevant doses of selegiline are far lower than concentrations of racemic or dextrorotatory methamphetamine that are known to be neurotoxic to dopaminergic neurons.[209][28] As such, dopaminergic neurotoxicity from selegiline's levomethamphetamine metabolite has been deemed unlikely.[28]
Newer formulations of selegiline, such as the ODT and transdermal patch forms, have been developed which strongly reduce formation of the amphetamine metabolites and their associated effects.[7][9] In addition, other MAO-B inhibitors that do not metabolize into amphetamines or monoamine releasing agents, like rasagiline and safinamide, have been developed and introduced.[38][212]
Dopaminergic neuroprotection
[edit]Starting around the age of 45, dopamine content in the caudate nucleus decreases at a rate of about 13% per decade, and this neurodegeneration extends to the nigrostriatal dopaminergic pathway in general.[20][32][213][133][214][215][216] This is a very high rate of neuronal decay relative to brain aging generally.[32] Similarly, age-related decay of mesolimbic dopaminergic neurons as well as noradrenergic neurons is substantially slower than in the nigrostriatal pathway.[32][215] Symptoms of Parkinson's disease are known to develop when the dopamine content of the caudate nucleus drops below 30% of the normal level.[32][213][214][133] Loss of striatal dopamine reaches a level of 40% in healthy people by the age of 75, whereas in people with Parkinson's disease, the loss is around 70% at diagnosis and more than 90% at death.[32] Only about 0.1% of the human population develops Parkinson's disease.[214][133][32] In these individuals, the nigrostriatal pathway deteriorates more rapidly and prematurely than usual, for instance at a rate of 30 to 90% loss of dopamine content per decade.[214][133] However, it is thought that if humans lived much longer than the average lifespan, everyone would eventually develop Parkinson's disease.[214][133] Besides the nigrostriatal pathway, there is also considerable, albeit lesser, loss of dopaminergic neurons in people with Parkinson's disease in other pathways and areas, like the mesolimbic and mesocortical pathways.[215] There is even substantial loss of dopamine in non-brain tissues, like the adrenal cortex and retina, implicating a generalized degeneration of the whole dopamine system.[215]
The progressive loss of dopaminergic neurons in the nigrostriatal pathway as well as other areas has implications not only for motor control and risk of Parkinson's disease but also for cognition, emotion, learning, sexual activity, and other processes.[20][32][215] Dopamine itself is thought to play a major role in this degeneration by metabolism into reactive oxygen species that damage dopaminergic neurons.[32] Age-related degeneration of nigrostriatal dopaminergic neurons is similar in rodents and humans.[32][213] Selegiline has been found to attenuate the age-related morphological changes in the nigrostriatal pathway of rodents and to produce accompanying preservations of cognitive and sexual functions.[20][32][213] These protective effects may be mediated by activities of selegiline including its MAO-B inhibition, its catetcholaminergic activity enhancer effects, and other actions.[20][32][213] According to József Knoll and Ildikó Miklya, two of the developers of selegiline, the drug may act as a neuroprotective and may be able to modestly slow the rate of age-related loss of dopamine signaling in humans.[20][32][161][163][217] Knoll has advocated for the widespread use of a low dose of selegiline (1 mg/day or 10–15 mg/week) in the healthy population for such purposes and has used this himself.[213][160][214][161][218] However, antiaging and anti-neurodegenerative effects of selegiline in humans have not been clearly demonstrated as of present and this theory remains to be substantiated.[50][20]
Other actions
[edit]Selegiline has a weak norepinephrine, dopamine, and serotonin-releasing effects, weakly blocks dopamine receptors, and weakly inhibits the reuptake of norepinephrine.[30][219][192] However, these actions are largely of very low potency and are of questionable clinical significance.[30] On the basis of positron emission tomography (PET) research with the ODT and patch formulations of selegiline, the drug does not significantly inhibit the brain dopamine transporter (DAT) in humans at clinical doses.[148]
Selegiline appears to activate σ1 receptors, having a relatively high affinity for these receptors of approximately 400 nM.[220][221]
Selegiline and its metabolite desmethylselegiline have been reported to directly bind to and inhibit glyceraldehyde-3-phosphate dehydrogenase (GAPDH).[38][222][223] This might play a modulating role in the clinical effectiveness of selegiline for Parkinson's disease.[38][222][223]
Unlike some of the hydrazine MAOIs like phenelzine and isocarboxazid, selegiline does not inhibit semicarbazide-sensitive amine oxidase (SSAO; also known as primary amine oxidase (PrAO) or as diamine oxidase (DAO)) nor does it pose a risk of vitamin B6 deficiency.[44] As a result, selegiline does not have risks of the side effects of these actions.[44]
Selegiline has been reported to inhibit several cytochrome P450 enzymes, including CYP2D6, CYP3A4/5, CYP2C19, CYP2B6, and CYP2A6.[8][123]
Pharmacokinetics
[edit]Absorption
[edit]Selegiline has an oral bioavailability of about 4 to 10%.[5][11][12][224] The average time to peak levels of selegiline is 0.6 to 1.4 hours in different studies, with a range of about 0.5 to 1.5 hours in one study.[5]
The circulating levels of selegiline and its metabolites following a single 10 mg oral dose have been studied.[5] The metabolites of selegiline include desmethylselegiline, levomethamphetamine, and levoamphetamine.[5] The average peak concentrations of selegiline across several studies ranged from 0.84 ± 0.6 μg/L to 2.2 ± 1.2 μg/L and the AUC levels ranged from 1.26 ± 1.19 μg⋅h/L to 2.17 ± 2.59 μg⋅h/L.[5] In the case of desmethylselegiline, the time to peak has been reported to be 0.8 ± 0.2 hours, the peak levels were 7.84 ± 2.11 μg/L to 13.4 ± 3.2 μg/L, and the area-under-the-curve (AUC) levels were 15.05 ± 4.37 μg⋅h/L to 40.3 ± 10.7 μg⋅h/L.[5] For levomethamphetamine, the peak levels were 10.2 ± 1.5 μg/L and the AUC levels were 150.2 ± 21.6 μg⋅h/L, whereas for levoamphetamine, the peak levels were 3.6 ± 2.9 μg/L and the AUC levels were 61.7 ± 44.0 μg⋅h/L.[5] For comparison, following a single 10 mg oral dose of dextromethamphetamine or dextroamphetamine, peak levels of these agents have been reported to range from 14 to 90 μg/L and from 15 to 34 μg/L, respectively.[225] Time to peak for levomethamphetamine has been reported to be 0.75 to 6 hours and for levoamphetamine has been reported to be 2.5 to 12 hours in people with different CYP2D6 metabolizer phenotypes.[40][114] Levels of desmethylselegiline, levomethamphetamine, and levoamphetamine are 4- to almost 20-fold higher than maximal selegiline levels with oral selegiline therapy.[115][4]
With repeated administration of selegiline, there is an accumulation of selegiline and its metabolites.[5] With a dosage of 10 mg once a day or 5 mg twice daily, peak levels of selegiline were 1.59 ± 0.89 μg/L to 2.33 ± 1.76 μg/L and AUC levels of selegiline were 6.92 ± 5.39 μg⋅h/L to 7.84 ± 5.43 μg⋅h/L after 1 week of treatment.[5] This equated to a 1.9- to 2.6-fold accumulation in peak levels and a 3.6- to 5.5-fold accumulation in AUC levels.[5] The metabolites of selegiline accumulate to a smaller extent than selegiline.[5] The AUC levels of desmethylselegiline increased by 1.5-fold and the peak and AUC levels of levomethamphetamine and levoamphetamine increased by 2-fold following 1 week of treatment with selegiline.[5] Selegiline appears to inhibit its own metabolism and that of desmethylselegiline with continuous use.[129][130]
The oral bioavailability of selegiline increases when it is ingested together with a fatty meal, as the molecule is fat-soluble.[3][226] There is a 3-fold increase in peak levels of selegiline and a 5-fold increase in AUC levels when it is taken orally with food.[5][4] The elimination half-life of selegiline is unchanged when it is taken with food.[5] In contrast to selegiline itself, the pharmacokinetics of its metabolites, desmethylselegililne, levomethamphetamine, and levoamphetamine, are unchanged when selegiline is taken with food.[5]
Distribution
[edit]The apparent volume of distribution of selegiline is 1,854 ± 824 L.[5] Selegiline and its metabolites rapidly cross the blood–brain barrier and enter the brain, where they are most concentrated in the thalamus, basal ganglia, midbrain, and cingulate gyrus.[54][8] Selegiline especially accumulates in brain areas with high MAO-B content, such as the thalamus, striatum, cortex, and brainstem.[25] Concentrations of selegiline's metabolites in cerebrospinal fluid (CSF) are similar to those in blood, suggesting that accumulation in the brain over peripheral tissues does not occur.[25]
No data were originally available on the plasma protein binding of selegiline.[5] It has been stated that the plasma protein binding of selegiline is 94%, but it has been said that there is no actual evidence to support this figure.[5] Subsequent research found that its plasma protein binding is 85 to 90%.[9][8][6]
Metabolism
[edit]Selegiline is metabolized in the intestines, liver, and other tissues.[5][25] More than 90% of orally administered selegiline is metabolized prior to reaching the bloodstream due to strong first-pass metabolism.[7] Selegiline (L-N-propargylmethamphetamine) is metabolized by N-demethylation into levomethamphetamine and by N-depropargylation into desmethylselegiline (L-N-propargylamphetamine).[9][44] Subsequently, levomethamphetamine is further metabolized into levoamphetamine by N-demethylation and desmethylselegiline is further metabolized into levoamphetamine by N-depropargylation.[7][44] Levomethamphetamine, levoamphetamine, and desmethylselegiline constitute the three major or primary metabolites of selegiline.[9][5][25] No racemization occurs in the metabolism of selegiline or its metabolites; that is, the levorotatory enantiomers are not converted into the dextrorotatory enantiomers, such as D-deprenyl, dextromethamphetamine, or dextroamphetamine.[11] Following their formation, the amphetamine metabolites of selegiline are also metabolized via hydroxylation and then conjugation via glucuronidation.[44] Besides the preceding metabolites, selegiline-N-oxide and formaldehyde are also known to be formed.[162] More than 40 minor metabolites of selegiline have been either detected or proposed.[162] Due to the amphetamine metabolites of selegiline, people taking selegiline may test positive for "amphetamine" or "methamphetamine" on drug screening tests.[227][228]
The exact cytochrome P450 enzymes responsible for the metabolism of selegiline have not been fully elucidated.[18] CYP2B6, CYP2C9, and CYP3A are thought to be significantly involved in the metabolism of selegiline on the basis of in vitro studies.[9][19][40] Other cytochrome P450 enzymes, including CYP1A2, CYP2A6, CYP2C8, CYP2D6, CYP2C19, and CYP2E1, may also be involved.[9][11][19][40] One review concluded that CYP2B6 and CYP2C19 are the leading candidates in selegiline metabolism.[18] CYP2B6 is thought to N-demethylate selegiline into desmethylselegiline and CYP2B6 and CYP2C19 are thought to N-depropargylate selegiline into levomethamphetamine.[9][19] Additionally, CYP2B6 and CYP2C19 are thought to metabolize desmethylselegiline into levoamphetamine and CYP2B6 is thought to N-demethylate levomethamphetamine into levoamphetamine.[9][19] CYP2D6 and CYP2C19 metabolizer phenotypes did not significantly affect the pharmacokinetics of selegiline, suggesting that these enzymes are minimally involved in its metabolism.[18][40][114][115] However, although most pharmacokinetic variables were unaffected, AUC levels of levomethamphetamine were 46% higher and its elimination half-life 33% longer in CYP2D6 poor metabolizers compared to extensive metabolizers and desmethylselegiline AUC levels were 68% higher in CYP2C19 poor metabolizers compared to extensive metabolizers.[40][114][115] As with CYP2D6 and CYP2C19, CYP3A4 and CYP3A5 are unlikely to be majorly involved in the metabolism of selegiline as the strong inhibitor itraconazole has minimal impact on its pharmacokinetics.[18][116][6]
Elimination
[edit]Selegiline administered orally is recovered 87% in urine and 15% in feces as the unchanged parent drug and its metabolites.[15][7][16] Of selegiline excreted in urine, 20 to 63% is excreted as levomethamphetamine, 9 to 26% as levoamphetamine, 1% as desmethylselegiline, and 0.01 to 0.03% at unchanged selegiline.[7][5][17] In the case of levomethamphetamine and levoamphetamine, with an oral dose of 10 mg selegiline, this would be amounts of about 2 to 6 mg levomethamphetamine and about 1 to 3 mg levoamphetamine.[7][17] The near-absence of unchanged excreted selegiline indicates that selegiline is essentially completely metabolized prior to its excretion.[5][7]
The average elimination half-life of selegiline after a single oral dose ranges from 1.2 to 1.9 hours across studies.[5] With repeated administration, the half-life of selegiline increases to 7.7 ± 12.6 hours to 9.6 ± 13.6 hours.[5] The elimination half-life of selegiline's metabolite, desmethylselegiline, has been reported to range from 2.2 ± 0.6 hours to 3.8 hours.[5] The half-lives of its metabolites levomethamphetamine and levoamphetamine have been reported to be 14 hours and 16 hours, respectively.[5] In another study, their half-lives were 11.6 to 15.4 hours and 17.0 to 18.1 hours, respectively, in people with different CYP2D6 metabolizer phenotypes.[40][114] Following repeated administration, the half-life of desmethylselegiline increased from 3.8 hours with the first dose to 9.5 hours following 1 week of daily selegiline doses.[5] Selegiline is a known inhibitor of several cytochrome P450 enzymes, such as CYP2B6 and CYP2A6.[124][123][125][129] It appears to inhibit its own metabolism and the metabolism of its metabolite desmethylselegiline.[129][130]
The oral clearance of selegiline is 59.4 ± 43.7 L/min.[5] This is described as very high and as almost 30-fold higher than hepatic blood flow.[5] The renal clearance of selegiline is 0.0072 L/h and is very low compared to its oral clearance.[5] These findings suggest that selegiline is extensively metabolized not only by the liver but also by non-hepatic tissues.[5]
Orally disintegrating tablet
[edit]Selegiline as an orally disintegrating tablet (ODT) is absorbed primarily buccally instead of being swallowed orally.[6][14] It was found to have 5- to 8-fold higher bioavailability, more consistent blood levels, and to produce fewer amphetamine metabolites than the standard oral tablet form.[14][13] It achieves blood levels of selegiline at a dose of 1.25 mg/day that are similar to those with conventional oral selegiline at a dose of 10 mg/day.[7] In addition, there is an at least 90% reduction in metabolites of selegiline including desmethylselegiline, levomethamphetamine, and levoamphetamine with the ODT formulation of selegiline compared to conventional oral selegiline.[7] Hence, levels of these metabolites are 10-fold lower with the ODT formulation.[187] The levels of amphetamine metabolites with the ODT formulation have been regarded as negligible.[6] This formulation of selegiline retains selectivity for MAO-B over MAO-A and likewise does not cause the "cheese effect" with consumption of tyramine-rich foods.[7]
Transdermal patch
[edit]The selegiline transdermal patch is indicated for application to the upper torso, upper thigh, or the outer upper arm once every 24 hours.[8] With application, an average of 25 to 30% (range 10 to 14%) of the selegiline content of the patch is delivered systemically over 24 hours.[9][8] This equates to about 0.3 mg selegiline per cm2 over 24 hours.[9] The patch has approximately 75% bioavailability, compared to 4 to 10% with the conventional oral form.[9][12] Transdermal selegiline results in significantly higher exposure to selegiline and lower exposure to all metabolites compared to conventional oral selegiline.[9] Selegiline levels are 50-fold higher and exposure to its metabolites 70% lower with the transdermal patch compared to oral administration at equivalent doses.[9] These differences are due to extensive first-pass metabolism with the oral form and the bypassing and absence of the first pass with the patch form.[9][12] Selegiline absorption and levels have been found to be equivalent when applied to the upper torso versus the upper thigh.[8] The drug does not accumulate in skin and is not significantly metabolized in skin.[8]
Hepatic and renal impairment
[edit]The United States drug label for oral selegiline states that no information is available on this formulation of the drug in the context of hepatic or renal impairment.[4] Conversely, the transdermal patch drug label states that no pharmacokinetic differences in selegiline and its metabolites were observed in mild or moderate liver impairment nor in mild, moderate, or severe renal impairment.[8] As such, the label states that dosage adjustment is not needed in these contexts.[8] Severe hepatic impairment and end-stage renal impairment were not studied.[8] In the case of the ODT formulation of selegiline, its drug label states that the dosage of selegiline should be reduced in mild and moderate hepatic impairment, whereas no dosage adjustment is required in mild to moderate renal impairment.[6] The label additionally states that ODT selegiline is not recommended in severe hepatic impairment nor in severe or end-stage renal impairment.[6] In clinical studies described by the ODT label, selegiline exposure was 1.5-fold higher and desmethylselegiline exposure 1.4-fold higher in mild hepatic impairment, selegiline exposure was 1.5-fold higher and desmethylselegiline exposure 1.8-fold higher in moderate hepatic impairment, and selegiline exposure was 4-fold higher and desmethylselegiline exposure 1.25-fold higher in severe hepatic impairment.[6] Conversely, levomethamphetamine and levoamphetamine exposures were not modified by hepatic impairment.[6] In the case of renal impairment, selegiline and desmethylselegiline levels were not substantially different in mild and moderate renal impairment and selegiline levels were likewise not substantially different in end-stage renal impairment.[6] However, levomethamphetamine and levoamphetamine exposures were increased by 34 to 67% in moderate renal impairment and by approximately 4-fold in end-stage renal impairment.[6]
In a published clinical study, hepatic and renal function were reported to more dramatically influence the pharmacokinetics of selegiline in the case of oral selegiline.[229][230][122] The pharmacokinetics of selegiline's major metabolites, desmethylselegiline, levomethamphetamine, and levoamphetamine, were also affected, but to a much lesser extent compared to selegiline itself.[122] AUC levels of selegiline relative to normal control subjects were 18-fold higher in people with hepatic impairment, 23-fold lower in people with drug-induced liver dysfunction, and 6-fold higher in people with renal impairment.[230][122] The drug-induced liver dysfunction group consisted of people taking a variety of anticonvulsants, including phenobarbital, phenytoin, carbamazepine, and amobarbital, that are known to strongly activate drug-metabolizing enzymes.[122] However, in a previous study, carbamazepine specifically did not reduce selegiline exposure.[8][9] Phenobarbital and certain other anticonvulsants are known to strongly induce CYP2B6, one of the major enzymes thought to be involved in the metabolism of selegiline, and it was concluded by the study authors that induction of this enzyme was the most likely explanation of the dramatically reduced exposure to selegiline in the drug-induced liver dysfunction group.[122] Because of these increased exposures, subsequent literature reviews citing the study have stated that selegiline (route/form not specified) is not recommended in people with moderate or severe liver impairment or with renal impairment.[119][231]
Chemistry
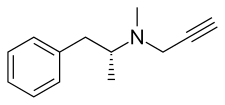
[edit]Selegiline is a substituted phenethylamine and amphetamine derivative.[40] It is also known as (R)-(–)-N,α-dimethyl-N-(2-propynyl)phenethylamine, (R)-(–)-N-methyl-N-2-propynylamphetamine, or N-propargyl-L-methamphetamine.[232][233][234][7] Selegiline (L-deprenyl) is the enantiopure levorotatory enantiomer of the racemic mixture deprenyl, whereas D-deprenyl is the dextrorotatory enantiomer.[41][20] Selegiline is a derivative of levomethamphetamine (L-methamphetamine), the levorotatory enantiomer of the psychostimulant and sympathomimetic agent methamphetamine (N-methylamphetamine), with a propargyl group attached to the nitrogen atom of the molecule.[61]
Selegiline is a small-molecule compound, with the molecular formula C13H17N and a low molecular weight of 187.281 g/mol.[232][233][234][4][61] It has high lipophilicity, with an experimental log P of 2.7 and predicted log P values of 2.9 to 3.1.[232][233][234][61] Pharmaceutically, selegiline is used almost always as the hydrochloride salt, though the free base form has also been used.[4][235] At room temperature, selegiline hydrochloride is a white to near white crystalline powder.[4] Selegiline hydrochloride is freely soluble in water, chloroform, and methanol.[4]
Analogues
[edit]Selegiline is a close analogue of methamphetamine and amphetamine, and in fact produces their levorotatory forms, levomethamphetamine and levoamphetamine, as metabolites.[38][28] Selegiline is structurally similar to the antihypertensive agent pargyline (N-methyl-N-propargylbenzylamine), an earlier non-selective MAOI of the phenylalkylamine group.[236][33] Besides selegiline and pargyline, another clinically used MAOI of the phenylalkylamine and amphetamine families is the antidepressant tranylcypromine (trans-2-phenylcyclopropylamine).[44] Tranylcypromine can be conceptualized as a cyclized amphetamine and has amphetamine-like actions at high doses similarly to selegiline.[44][237][238] Another notable analogue of selegiline is 4-fluoroselegiline, a variation of selegiline in which one of the hydrogen atoms of the phenyl ring has been replaced with a fluorine atom.[239] A large number of other analogues of selegiline derived via structural modification have been synthesized and characterized.[240][239][241][242]
Rasagiline (N-propargyl-1(R)-aminoindan) is an analogue of selegiline in which the amphetamine base structure has been replaced with a 1-aminoindan structure and the N-methyl group has been removed.[38] Like selegiline, it is also a selective MAO-B inhibitor and used to treat Parkinson's disease.[38] In contrast to selegiline however, rasagiline lacks the amphetamine metabolites and activity of selegiline.[38] A further derivative of rasagiline, ladostigil ([N-propargyl-(3R)-aminoindan-5-yl]-N-propylcarbamate), a dual MAO-B inhibitor and acetylcholinesterase inhibitor, was developed for treatment of Alzheimer's disease and other conditions but was ultimately never introduced for medical use.[243]
Synthesis
[edit]Selegiline can be synthesized by the alkylation of levomethamphetamine using propargyl bromide.[44][244][245][246][247]

History
[edit]Following the discovery in 1952 that the tuberculosis drug iproniazid elevated the mood of people taking it, and the subsequent discovery that the effect was likely due to inhibition of monoamine oxidase (MAO) and elevation of monoamine neurotransmitters in the brain, many people and companies started trying to discover monoamine oxidase inhibitors (MAOIs) to use as antidepressants.[11][248] Deprenyl, the racemic form of selegiline, was synthesized and discovered by Zoltan Ecseri at the Chinoin Pharmaceutical Company (part of Sanofi since 1993) in Budapest, Hungary.[11][249] Chinoin received a patent on the drug in 1962 and the compound was first published in the scientific literature in English in 1965.[11][250] Chinoin researchers had been studying substituted amphetamines since 1960, and decided to try synthesizing amphetamines that acted as MAOIs.[65] It had been known that methamphetamine was a reversible inhibitor of MAO.[65] Deprenyl, also known as N-propargyl-N-methylamphetamine,[33] is closely related to and inspired by pargyline (N-propargyl-N-methylbenzylamine), another MAOI that had been synthesized earlier.[11][65][251] Deprenyl was initially referred to by the chemical name phenylisopropylmethylpropinylamine and the developmental code name E-250.[11][250] Work on the biology and effects of E-250 in animals and humans was conducted by a group led by József Knoll at Semmelweis University, which was also in Budapest.[11]
Deprenyl is a racemic compound (a mixture of two isomers called enantiomers).[11][65] Further work determined that the levorotatory enantiomer was a more potent MAOI, which was published in 1967, and subsequent work was done with the single enantiomer L-deprenyl.[11][65][154][218] In 1968, it was discovered by Johnston that monoamine oxidase exists in multiple forms.[11][65][252] In 1971, Knoll showed that selegiline highly selectively inhibits the B-isoform of monoamine oxidase (MAO-B) and proposed that it is unlikely to cause the infamous "cheese effect" (hypertensive crisis resulting from consuming foods containing tyramine) that occurs with non-selective MAOIs.[11][65][253] The lack of potentiation of tyramine effect by deprenyl had previously been reported in 1966 and 1968 studies, but could not be mechanistically explained until after the existence of multiple forms of MAO was discovered.[11][65][254] Selegiline was the first selective MAO-B inhibitor to be discovered[13] and is described as prototypical of these agents.[44][45]
Deprenyl and selegiline were initially studied as antidepressants for treatment of depression.[160][250] Deprenyl was first found to be effective for depression from 1965 to 1967,[160][255][256] while selegiline was first found to be effective for depression in 1971 and this was further corroborated in 1980.[160][257][258] A 1984 study that combined selegiline with phenylalanine reported remarkably high effectiveness in the treatment of depression similar to that with electroconvulsive therapy (ECT).[160][259] However, selegiline in its original oral form was never further developed or approved for the treatment of depression.[160]
A few years after the discovery that selegiline was a selective MAO-B inhibitor, two Parkinson's disease researchers based in Vienna, Peter Riederer and Walther Birkmayer, realized that selegiline could be useful in Parkinson's disease. One of their colleagues, Moussa B. H. Youdim, visited Knoll in Budapest and took selegiline from him to Vienna. In 1975, Birkmayer's group published the first paper on the effect of selegiline in Parkinson's disease.[218][260]
Speculation that selegiline could be useful as an anti-aging drug or aphrodisiac based on animal studies began in the 1970s.[261]
Selegiline was first introduced for clinical use in Hungary in 1977.[42] It was approved in the oral pill form under the brand name Jumex to treat Parkinson's disease.[42] The drug was then introduced in the United Kingdom in 1982.[42] In 1987, Somerset Pharmaceuticals in New Jersey, which had acquired the rights to develop selegiline in the United States, filed a New Drug Application (NDA) with the Food and Drug Administration (FDA) to market the drug for Parkinson's disease in this country.[43] While the NDA was under review, Somerset was acquired in a joint venture by two generic drug companies, Mylan and Bolan Pharmaceuticals.[43] Selegiline was approved for Parkinson's disease by the FDA in 1989.[43]
It had been known since the mid-1960s that high doses of deprenyl had psychostimulant effects.[30][11][250][256] Selegiline was first shown to metabolize into levomethamphetamine and levoamphetamine in humans in 1978.[28][262] The involvement of these metabolites in the effects and side effects of selegiline has remained controversial and unresolved in the decades afterwards.[28][38] In any case, concerns about these metabolites have contributed to the development of newer MAO-B inhibitors like rasagiline and safinamide that lack such metabolites.[38][212]
The catecholaminergic activity enhancer (CAE) effects of selegiline became well-characterized and distinctly named in 1994.[159][32][25][178][20][34][177][176][263] These effects had been observed much earlier, dating back to the 1960s and 1970s, but were not properly distinguished from the other actions of selegiline, like MAO-B inhibition, until the 1990s.[32][25][34][159] More potent, selective, and/or expansive monoamine activity enhancers (MAEs), like phenylpropylaminopentane (PPAP) and benzofuranylpropylaminopentane (BPAP), were derived from selegiline and other compounds and were first described in 1992 and 1999, respectively.[33][36][171][160] These drugs had been proposed for potential treatment of psychiatric disorders like depression as well as for Parkinson's disease and Alzheimer's disease, but were never developed or marketed.[264][34][36][163][160]
In the 1990s, J. Alexander Bodkin at McLean Hospital, an affiliate of Harvard Medical School, began a collaboration with Somerset to develop delivery of selegiline via a transdermal patch in order to avoid the well known dietary restrictions of MAOIs.[261][265][266] Somerset obtained FDA approval to market the patch for depression in 2006.[267] Similarly, the orally disintegrating tablet (ODT) form of selegiline, marketed under the brand name Zelapar, was approved for Parkinson's disease in the United States in 2006 and in the European Union in 2010.[42]
Binding to and agonism of the trace amine-associated receptors (TAARs) as the mechanism responsible for the MAE effects of selegiline and related MAEs like PPAP and BPAP was first suggested in the early 2000s following the discovery of the TAARs.[36][163][37] Activation of the TAAR1 as the mechanism of the MAE effects was first clearly substantiated in 2022.[167][35] TAAR1 agonists like ulotaront and ralmitaront are under development for treatment of various psychiatric disorders as of 2023.[180][181]
Society and culture
[edit]Names
[edit]Selegiline is the generic name of the drug and its INN, BAN, and DCF, while selegiline hydrochloride is the USAN.[268][269][235] The word "selegiline" is pronounced /səˈlɛdʒɪliːn/ (sə-LEJ-i-leen) or as "seh-LEH-ji-leen".[1][2] Selegiline is also known as L-deprenyl, L-deprenil, L-deprenalin, L-deprenaline, L-phenylisopropylmethylpropinylamine, and L-E-250.[20][268][269][235][250] It should not be confused with the racemic form, deprenyl (E-250), or with the dextrorotatory enantiomer, D-deprenyl, which are distinct substances.[268][41][20]
Major brand names of selegiline include Eldepryl, Jumex, and Movergan (oral tablet and/or capsule), Zelapar (orally disintegrating tablet or ODT), and Emsam (transdermal patch).[3][235][162] Selegiline has been marketed under more than 70 brand names worldwide.[165][3] The brand name "Emsam" was derived from the names of two children, Emily and Samuel, of one of the executives at Somerset Pharmaceuticals, the developer of Emsam.[61][270]
Generic forms
[edit]Generic forms of oral selegiline are available in the United States.[51] However, generic forms of the orally disintegrating tablet and the transdermal patch are not available in this country.[51][52] The latter formulations of selegiline are very expensive, and this can be prohibitive to their use.[52][271] There has been poor insurance coverage of the transdermal patch form for depression, with insurance companies often requiring patients to first fail to respond to one or two other antidepressants and to be responsible for larger copayments.[52] It is expected that generics of the transdermal patch will become available at some point in the future.[52]
Availability
[edit]Conventional oral selegiline (brand names Eldepryl, Jumex) is widely marketed throughout the world, including in over 70 countries.[3][235][20][165] Conversely, the selegiline transdermal patch (brand name Emsam) is only marketed in the United States, while the selegiline orally disintegrating tablet (brand name Zelapar) is marketed in the United States, the United Kingdom, and the European Union.[3][42][20]
Notable users
[edit]József Knoll, one of the developers of selegiline, began taking a low 1 mg daily dose of selegiline on January 1, 1989 at the age of 64.[161]: 92 [218] He reported in 2012 that this had continued for 22 years uninterrupted.[161]: 92 Knoll stated that he had become so fascinated with the possible longevity-promoting effects of selegiline that he had decided to start taking it as a self-experiment.[161]: 92 [218] Knoll later died in 2018 at the age of 93.[272]
David Pearce, a British transhumanist philosopher, wrote his self-published book-length internet manifesto The Hedonistic Imperative[273] six weeks after starting to take selegiline.[274]
Sam Bankman-Fried, the founder and former CEO of the FTX cryptocurrency exchange, is known to have used selegiline for depression in the form of the Emsam patch for at least 5 to 10 years.[275][276] He is also known to have simultaneously taken Adderall for treatment of attention deficit hyperactivity disorder (ADHD)[275][276] and to have possessed non-pharmaceutical adrafinil, a prodrug of modafinil.[277]
Fictional representations
[edit]In Gregg Hurwitz's novel Out of the Dark, selegiline (Emsam) and tyramine-containing food were used to assassinate the president of the United States.[278]
Internet vendors
[edit]Selegiline in non-pharmaceutical form is sold on the Internet without a prescription by online vendors for uses such as purported cognitive enhancement (i.e., as a so-called "smart drug" or nootropic) and anti-aging effects.[279][178][280] It is widely available for such purposes, for instance under informal brand names like Dep-Pro, Selepryl, and Cyprenil, which are oral liquid solutions of selegiline at a concentration of 1 mg per drop.[178][280][161]: 86
Presence in ecstasy
[edit]In his 1993 book E for Ecstasy examining the uses of the street drug ecstasy in the United Kingdom, the writer, activist, and ecstasy advocate Nicholas Saunders highlighted test results showing that certain consignments of the drug also contained selegiline.[281] Consignments of ecstasy known as "Strawberry" contained what Saunders described as a "potentially dangerous combination of ketamine, ephedrine and selegiline," as did a consignment of "Sitting Duck" Ecstasy tablets.[282]
Doping in sport
[edit]Selegline is on the World Anti-Doping Agency (WADA)'s list of prohibited substances.[283] It is classified as a "stimulant" in this list, along with various amphetamines, methylphenidate, adrenergic sympathomimetics, modafinil, and other agents.[283] A review of the pharmacology of WADA prohibited substances noted that although selegiline is classified as a stimulant in the WADA prohibited substances list and stimulants can enhance physical performance, selegiline was seemingly included in the list not because of any short-term stimulant effects of its own, but rather because it metabolizes into small amounts of levomethamphetamine and levoamphetamine and can produce false positives for amphetamines on drug tests.[283] In any case, levomethamphetamine and levoamphetamine are catecholamine releasing agents and can produce sympathomimetic and psychostimulant effects with sufficiently high exposure.[186][203][202] Such actions may have performance-enhancing effects.[283]
Regulatory status
[edit]Selegiline is a prescription drug.[4][8][6] It is not specifically a controlled substance in the United States and hence is not an illegal drug.[8] However, deprenyl and selegiline are controlled substances in Japan.[284][285] They are classified as "Stimulants", alongside a variety of other amphetamines, under Article 2 of Japan's Narcotics and Psychotropics Control Law.[285] Selegiline is known to metabolize into small amounts of levoamphetamine and levomethamphetamine but is thought to have little to no misuse potential or dependence liability.[26][27][28][29][85][8]
Non-medical use
[edit]Anti-aging and longevity
[edit]József Knoll and his team are credited with having developed selegiline. Although selegiline's development as a potential treatment for Parkinson's disease, Alzheimer's disease, and depression was headed by other teams, Knoll remained at the forefront of research into the potential longevity enhancing effects of selegiline up until his death in 2018.[272][286][287] Knoll published his 2012 book How Selegiline ((–)-Deprenyl) Slows Brain Aging wherein he claims that:[161]: 90
"In humans, maintenance from sexual maturity on (–)-deprenyl (1mg daily) is, for the time being, the most promising prophylactic treatment to fight against the age related decay of behavioral performances, prolonging life, and preventing or delaying the onset of age-related neurodegenerative diseases such as Parkinson's and Alzheimer's".
The mechanism of selegiline's longevity-promoting effect has been researched by several groups, including Knoll and his associates at Semmelweis University, Budapest.[20] The drug has been determined to be a catecholaminergic activity enhancer when present in minuscule concentrations far below those at which monoamine oxidase inhibitory activity can be observed, thereby potentiating the release of catecholamine neurotransmitters in response to stimuli. Knoll maintains that micro-doses of selegiline act as a synthetic analogue to a known or unknown trace amine in order to preserve the brain catecholaminergic system, which he perceives as integral to the organism's ability to function in an adaptive, goal-directed and motivated manner during advancing physical age:[161]: 70, 43
"[...] enhancer regulation in the catecholaminergic brain stem neurons play[s] a key role in controlling the uphill period of life and the transition from adolescence to adulthood. The results of our longevity studies support the hypothesis that quality and duration of life rests upon the inborn efficiency of the catecholaminergic brain machinery, i.e. a high performing, long-living individual has a more active, more slowly deteriorating catecholaminergic system than its low performing, shorter living peer. Thus, a better brain engine allows for a better performance and a longer lifespan."
"Since the catecholaminergic and serotonergic neurons in the brain stem are of key importance in ensuring that the mammalian organism works as a purposeful, motivated, goal-directed entity, it is hard to overestimate the significance of finding safe and efficient means to slow the decay of these systems with passing time. The conclusion that the maintenance on (–)-deprenyl that keeps the catecholaminergic neurons on a higher activity level is a safe and efficient anti-aging therapy follows from the discovery of the enhancer regulation in the catecholaminergic neurons of the brain stem. From the finding that this regulation starts working on a high activity level after weaning and the enhanced activity subsists during the uphill period of life, until sexual hormones dampen the enhancer regulation in the catecholaminergic and serotonergic neurons in the brain stem, and this event signifies the transition from developmental longevity into postdevelopmental longevity, the downhill period of life."
Despite findings by Knoll that selegiline can prolong lifespan in rodents by 35% however, other studies have had conflicting findings and have even found increased mortality with selegiline in rodents.[50] In humans with Parkinson's disease, selegiline has been associated with cardiovascular and psychiatric complications and has not been found to reduce mortality in long-term studies.[50] As such, the claimed anti-aging and longevity benefits of selegiline are controversial and uncertain.[50][49]
Nootropic or "smart drug"
[edit]Selegiline is considered by some to be a nootropic, otherwise known as a cognitive enhancer or "smart drug", both at clinical and sub-clinical dosages, and has been used off-label and non-medically to improve cognitive performance.[47][288] It is one of the most popular such agents.[47] Selegiline has been found to have neuroprotective activity against certain neurotoxins and to increase the production of several brain growth factors, such as nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF), and glial cell line-derived neurotrophic factor (GDNF).[20] The drug has also been found in animal models to improve learning ability and to help preserve it during ischemia and aging.[289][290][291][133] Despite claims that selegiline and other claimed nootropics have cogintive-enhancing effects however, these effects are controversial and their benefits versus risks are uncertain.[47]
Research
[edit]Depression
[edit]Selegiline has been clinically studied in combination with oral L-phenylalanine or β-phenethylamine in the treatment of depression and was reported to be effective.[36][151][259][292][293] L-Phenylalanine is known to be metabolized into β-phenethylamine, selegiline is known to strongly inhibit the metabolism of β-phenethylamine, and β-phenethylamine has been implicated in having psychostimulant-like mood-lifting effects.[36][30][151]
Social anxiety
[edit]A small clinical study found that oral selegiline (10 mg/day) reduced symptoms of social anxiety disorder.[12][21][294] The effectiveness was modest, with a reduction in social anxiety scores from baseline of 32% over 6 weeks of treatment.[12][21][294] It was seemingly less effective than certain other agents used in the treatment of social anxiety, such as the non-selective MAOI phenelzine (45% symptom reduction) and the benzodiazepine clonazepam (51% symptom reduction), though it was similar to the SSRI sertraline (32% symptom decrease).[294]
ADHD
[edit]Selegiline has been limitedly studied in the treatment of attention deficit hyperactivity disorder (ADHD) in children, adolescents, and adults.[21][295][296][297] In a small randomized trial of selegiline for treatment of ADHD in children, there were improvements in attention, hyperactivity, and learning/memory performance but not in impulsivity.[298] A small clinical randomized trial compared selegiline to methylphenidate, a first line treatment for ADHD, and reported equivalent efficacy as assessed by parent and teacher ratings.[299] In another small randomized controlled trial of selegiline for the treatment of adult ADHD, a high dose of the medication for 6 weeks was not significantly more effective than placebo in improving symptoms.[296][300][301] Selegiline in its transdermal patch form (brand name Emsam) has also been assessed in the treatment of ADHD in children and adolescents in a small open-label pilot study sponsored by the manufacturer in 2003.[12][302] However, there was a high rate of discontinuation and development was not further pursued.[12][302]
Motivational disorders
[edit]Selegiline has been found to increase effort expenditure and to reverse pharmacologically-induced motivational deficits in rodents.[303][304][305][306] In case reports and small clinical studies, selegiline has been reported to improve apathy in people with traumatic brain injury, stroke, and schizophrenia.[303][307][308] In accordance with the preceding findings, selegiline, along with other dopaminergic and activating agents, is a potentially promising treatment for disorders of diminished motivation, including apathy, abulia, and akinetic mutism.[304][309][308]
Addiction
[edit]Selegiline has been evaluated for smoking cessation both as a monotherapy and in combination with nicotine replacement therapy in five clinical studies.[310][311][21] However, it is limitedly or not effective for this use.[310][311][21] It was also evaluated for treatment of cocaine dependence in one study, but was similarly not effective.[312] Studies are mixed on whether selegiline, at MAO-B-selective doses, reduces the effects of cocaine in humans.[98][99][100][101][102][103] Selegiline, also at an MAO-B-selective dosage, did not modify or potentiate the pharmacological effects of intravenous methamphetamine in a small clinical study.[96][97]
Sexual dysfunction
[edit]Selegiline has been assessed for treatment of sexual dysfunction induced by antipsychotics in people with schizophrenia, but was not effective in a single small clinical study.[313][314] It also did not improve sexual function in men with depression, but did improve several domains of sexual function in women with depression.[63]
Psychosis
[edit]Selegiline has been studied as an adjunct to antipsychotics in the treatment of schizophrenia in four clinical studies.[21][315] However, it failed to significantly reduce positive or negative symptoms of schizophrenia in meta-analyses of these studies.[21][315]
Excessive sleepiness
[edit]Selegiline has been evaluated for the treatment of narcolepsy in three small clinical studies.[316][197][317] It was found to be effective in these studies.[316][197] A dosage of 10 mg/day had no effect on symptoms, but 20 to 30 mg/day improved alertness, mood, and somewhat reduced cataplexy, clinical effects that have been described as comparable to the same dosages of amphetamine.[197] Animal research indicates that the beneficial effects of high doses of selegiline in narcolepsy are likely due to conversion into its active metabolites, levoamphetamine and levomethamphetamine.[197][317] Selegiline has also been evaluated for treatment of hypersomnia (excessive sleeping or sleepiness) in people with myotonic dystrophy, but was not effective in a single small clinical study.[318][316]
Periodic limb movement disorder
[edit]Selegiline has been studied in the treatment of periodic limb movement disorder (PLMD) in a single small open-label clinical study.[319][320][321] It was reported to be effective as assessed by polysomnography, reducing periodic limb movements during sleep by about 60%.[319][321] Selegiline has not been studied for the related condition restless legs syndrome (RLS) as of 2023.[319][320] The drug has not been studied well enough in PLMD or RLS to be widely used in their treatment.[319]
Tardive dyskinedia
[edit]Selegiline was studied in the treatment of antipsychotic-induced tardive dyskinesia in one small clinical study, but was ineffective.[322]
Dementia and stroke
[edit]Selegiline has also been used off-label as a palliative treatment for dementia in Alzheimer's disease.[54] However, its clinical effectiveness is limited or lacking for this use.[323][134][324][325] It was also ineffective in the treatment of Lewy body dementia.[326] Selegiline has been used to support motor rehabilitation in stroke recovery, but evidence for this use is inadequate and no recommendation can be made for or against it.[327]
Disorders of consciousness
[edit]Selegiline has been studied in patients with disorders of consciousness, such as minimally conscious state, persistent vegetative state, and persistent coma, in a small open-label clinical study.[328][329] It was found to be effective in enhancing arousal and promoting recovery of consciousness in some of these individuals.[328][329]
Neurotoxicity
[edit]Selegiline has been reported to protect against the damage caused by the potent dopaminergic and/or noradrenergic neurotoxins 6-hydroxydopamine (6-OHDA), N-(2-chloroethyl)-N-ethyl-2-bromobenzylamine (DSP-4), and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) in animals.[330][30][331][332][333][334] Conversely, selegiline is ineffective in protecting against the serotonergic and noradrenergic neurotoxin 5,7-dihydroxytryptamine (5,7-DHT).[30][335]
Selegiline has also been reported to protect against methylenedioxymethamphetamine (MDMA)-induced serotonergic neurotoxicity in rodents.[336][337][338][339][340] The serotonergic neurotoxicity of MDMA appears to be dependent on release of dopamine and its subsequent metabolism by MAO-B within serotonergic neurons into hydroxyl radicals, which is blocked by MAO-B inhibition.[336][337] Likewise, selegiline prevented the serotonergic neurotoxicity of a combination of methylenedioxyaminoindane (MDAI) and dextroamphetamine.[341][342]
Conversely, selegiline failed to reduce the serotonergic neurotoxicity caused by fenfluramine and either did not affect or potentiated the serotonergic neurotoxicity caused by para-chloroamphetamine (PCA).[332][343][344][345] In addition, findings are mixed and conflicting on whether selegiline prevents amphetamine- and methamphetamine-induced dopaminergic neurotoxicity in rodents.[346][347][348][349]
Although MAO-B-selective doses of selegiline protect against MDMA-induced serotonergic neurotoxicity in rodents, combination of amphetamines like MDMA with MAOIs, including selegiline, can produce serious complications, including serotonin syndrome, hypertensive crisis, and death.[350][351]
Other formulations
[edit]The original oral formulation of selegiline was developed for the treatment of depression.[160] However, it ended up being developed and approved for the treatment of Parkinson's disease instead.[160][42][4] In any case, oral selegiline has been widely used off-label to treat depression.[21] The transdermal patch form of selegiline was developed and approved specifically for the treatment of depression.[352][12][9][8] It was also under development for the treatment of Alzheimer's disease, attention deficit hyperactivity disorder (ADHD), cognition disorders, and Parkinson's disease, but development for these indications was discontinued.[352] The ODT form of selegiline was developed and licensed exclusively for the treatment of Parkinson's disease.[353][7][6]
Veterinary use
[edit]In veterinary medicine, selegiline is sold under the brand name Anipryl and is manufactured by Zoetis.[354] It is available in the form of 2, 5, 10, 15, and 30 mg oral tablets for use in animals.[354] Selegiline is used in dogs to treat canine cognitive dysfunction (CCD) and, at higher doses, to treat pituitary-dependent hyperadrenocorticism (PDH).[355][356]
CCD is a form of dementia that mimics Alzheimer's disease in humans.[357] Geriatric dogs treated with selegiline show improvements in sleeping pattern, reduced incontinence, and increased activity level, with most showing improvements by one month of treatment.[358][359] Though it is labeled for use in dogs only, selegiline has been used off-label for geriatric cats with cognitive dysfunction.[360]
PDH is a hormonal disorder and is analogous to pituitary-dependent Cushing's syndrome in humans.[354] Selegiline's effectiveness in treating PDH has been disputed.[355] Theoretically, it works by increasing dopamine levels, which downregulates the secretion of adrenocorticotropic hormone (ACTH) from the brain, eventually leading to reduced levels of cortisol.[360] Some claim that selegiline is only effective at treating PDH caused by lesions in the anterior pituitary (which comprise most canine cases).[361] The greatest sign of improvement is lessening of PDH-related abdominal distention.[358]
Side effects in dogs are uncommon, but they include vomiting, diarrhea, diminished hearing, salivation, decreased weight, and behavioral changes such as hyperactivity, listlessness, disorientation, and repetitive motions.[356][361]
Selegiline has been limitedly studied in large animals like horses and its dosage in these animals has not been established.[361] In preliminary research, a dose of selegiline of 30 mg orally or intravenously in horses had no observable effects on behavior or locomotor activity.[361]
The doses of selegiline used in animals are described as extremely high relative to those used in humans (which are ~0.1 mg/kg body weight).[162]
Notes
[edit]- ^ Selegiline given subcutaneously to rodents selectively inhibits MAO-B with a single 0.2–2.0 mg/kg dose or a continuous 0.05 to 0.25 mg/kg dosage and substantially inhibits MAO-A at a continuous dosage of 1.0 mg/kg.[25] It also produces catecholaminergic activity enhancer (CAE) effects with a subcutaneous dose of 0.01 mg/kg (+42% activity) for noradrenergic neurons and at a dose of 0.025 mg/kg (+17% activity) for dopaminergic neurons.[25] For comparison, the dosage used in humans orally is around 1 mg per 10 kg or 0.1 mg/kg daily.[162][25]
- ^ Smith & Davis (1977) reviewed 11 clinical studies of dextroamphetamine and levoamphetamine including doses and potency ratios in terms of a variety of psychological and behavioral effects.[202] The summaries of these studies are in Table 1 of the paper.[202]
References
[edit]- ^ Jump up to: a b "Drug Treatments for Parkinson's" (PDF). Retrieved July 5, 2024.
Selegiline (seh-LEH-ji-leen)
- ^ Jump up to: a b Acosta WR (2020). Pharmacology for Health Professionals. Jones & Bartlett Learning. p. 66. ISBN 978-1-284-24083-2. Retrieved July 5, 2024.
sell-eh'-geh-leen
- ^ Jump up to: a b c d e f g h "Selegiline". Drugs.com. Archived from the original on July 3, 2024. Retrieved February 7, 2016.
- ^ Jump up to: a b c d e f g h i j k l m n o p q r s t u v w x y z aa ab ac ad "ELDEPRYL® (Selegiline Hydrochloride) Tablets, USP Label" (PDF). Food and Drug Administration. January 2008. Retrieved July 3, 2024.
- ^ Jump up to: a b c d e f g h i j k l m n o p q r s t u v w x y z aa ab ac ad ae af ag ah ai aj ak al am an ao ap aq ar as at au av aw ax ay az ba bb bc bd be bf bg bh bi bj bk bl bm bn Mahmood I (August 1997). "Clinical pharmacokinetics and pharmacodynamics of selegiline. An update". Clin Pharmacokinet. 33 (2): 91–102. doi:10.2165/00003088-199733020-00002. PMID 9260033.
- ^ Jump up to: a b c d e f g h i j k l m n o p q r s t u v w x y z aa ab ac ad ae af ag ah ai aj "ZELAPAR® (Selegiline Hydrochloride) Orally Disintegrating Tablets" (PDF). Food and Drug Administration. July 2021. Retrieved July 3, 2024.
- ^ Jump up to: a b c d e f g h i j k l m n o p q r s t u v w x y z aa ab Poston KL, Waters C (October 2007). "Zydis selegiline in the management of Parkinson's disease". Expert Opin Pharmacother. 8 (15): 2615–2624. doi:10.1517/14656566.8.15.2615. PMID 17931095.
- ^ Jump up to: a b c d e f g h i j k l m n o p q r s t u v w x y z aa ab ac ad ae af ag ah ai aj ak al am an ao ap aq ar as at au av "EMSAM® (Selegiline Transdermal System) Label" (PDF). Food and Drug Administration. July 2017. Retrieved July 2, 2024.
- ^ Jump up to: a b c d e f g h i j k l m n o p q r s t u v w x y z aa ab ac ad ae af ag ah ai aj ak Lee KC, Chen JJ (November 2007). "Transdermal selegiline for the treatment of major depressive disorder". Neuropsychiatric Disease and Treatment. 3 (5): 527–537. doi:10.2147/ndt.s12160200 (inactive July 6, 2024). PMC 2656289. PMID 19300583.
{{cite journal}}: CS1 maint: DOI inactive as of July 2024 (link) - ^ Anvisa (March 31, 2023). "RDC Nº 784 - Listas de Substâncias Entorpecentes, Psicotrópicas, Precursoras e Outras sob Controle Especial" [Collegiate Board Resolution No. 784 - Lists of Narcotic, Psychotropic, Precursor, and Other Substances under Special Control] (in Brazilian Portuguese). Diário Oficial da União (published April 4, 2023). Archived from the original on August 3, 2023. Retrieved August 16, 2023.
- ^ Jump up to: a b c d e f g h i j k l m n o p q r s t u v w x y z aa ab ac Magyar K (2011). "The Pharmacology of Selegiline". In Youdim M, Riederer P (eds.). Monoamine Oxidases and Their Inhibitors. International Review of Neurobiology. Vol. 100. Academic Press. pp. 65–84. doi:10.1016/B978-0-12-386467-3.00004-2. ISBN 978-0-12-386467-3. PMID 21971003.
- ^ Jump up to: a b c d e f g h i j k l m n Pae CU, Lim HK, Han C, Neena A, Lee C, Patkar AA (August 2007). "Selegiline transdermal system: current awareness and promise". Prog Neuropsychopharmacol Biol Psychiatry. 31 (6): 1153–1163. doi:10.1016/j.pnpbp.2007.04.020. PMID 17614182.
- ^ Jump up to: a b c d e Löhle M, Storch A (November 2008). "Orally disintegrating selegiline for the treatment of Parkinson's disease". Expert Opin Pharmacother. 9 (16): 2881–2891. doi:10.1517/14656566.9.16.2881. PMID 18937619.
- ^ Jump up to: a b c Clarke A, Brewer F, Johnson ES, Mallard N, Hartig F, Taylor S, et al. (November 2003). "A new formulation of selegiline: improved bioavailability and selectivity for MAO-B inhibition". Journal of Neural Transmission. 110 (11): 1241–1255. doi:10.1007/s00702-003-0036-4. PMID 14628189. S2CID 711419.
- ^ Jump up to: a b c Heinonen EH, Anttila MI, Lammintausta RA (December 1994). "Pharmacokinetic aspects of l-deprenyl (selegiline) and its metabolites". Clin Pharmacol Ther. 56 (6 Pt 2): 742–749. doi:10.1038/clpt.1994.204. PMID 7995016.
- ^ Jump up to: a b Heinonen EH, Myllylä V, Sotaniemi K, Lamintausta R, Salonen JS, Anttila M, et al. (November 1989). "Pharmacokinetics and metabolism of selegiline". Acta Neurologica Scandinavica. Supplementum. 126: 93–99. doi:10.1111/j.1600-0404.1989.tb01788.x. PMID 2515726. S2CID 221440315.
- ^ Jump up to: a b c d e f g Chrisp P, Mammen GJ, Sorkin EM (May 1991). "Selegiline: A Review of its Pharmacology, Symptomatic Benefits and Protective Potential in Parkinson's Disease". Drugs Aging. 1 (3): 228–248. doi:10.2165/00002512-199101030-00006. PMID 1794016.
- ^ Jump up to: a b c d e f g h i j k l m n o Rodrigues AD (June 2022). "Drug Interactions Involving 17α-Ethinylestradiol: Considerations Beyond Cytochrome P450 3A Induction and Inhibition". Clin Pharmacol Ther. 111 (6): 1212–1221. doi:10.1002/cpt.2383. PMID 34342002.
- ^ Jump up to: a b c d e f Hidestrand M, Oscarson M, Salonen JS, Nyman L, Pelkonen O, Turpeinen M, et al. (November 2001). "CYP2B6 and CYP2C19 as the major enzymes responsible for the metabolism of selegiline, a drug used in the treatment of Parkinson's disease, as revealed from experiments with recombinant enzymes". Drug Metab Dispos. 29 (11): 1480–1484. PMID 11602525.
- ^ Jump up to: a b c d e f g h i j k l m n o p q r s t u v w Miklya I (November 2016). "The significance of selegiline/(-)-deprenyl after 50 years in research and therapy (1965-2015)". Molecular Psychiatry. 21 (11): 1499–1503. doi:10.1038/mp.2016.127. PMID 27480491.
- ^ Jump up to: a b c d e f g h i j k l m n o p q r s t u v w x y Rossano F, Caiazza C, Sobrino A, Solini N, Vellucci A, Zotti N, et al. (July 2023). "Efficacy and safety of selegiline across different psychiatric disorders: A systematic review and meta-analysis of oral and transdermal formulations". Eur Neuropsychopharmacol. 72: 60–78. doi:10.1016/j.euroneuro.2023.03.012. PMID 37087864.
- ^ Jump up to: a b c d e f g h i j k l Citrome L, Goldberg JF, Portland KB (November 2013). "Placing transdermal selegiline for major depressive disorder into clinical context: number needed to treat, number needed to harm, and likelihood to be helped or harmed". Journal of Affective Disorders. 151 (2): 409–417. doi:10.1016/j.jad.2013.06.027. PMID 23890583.
- ^ Jump up to: a b Cipriani A, Furukawa TA, Salanti G, Chaimani A, Atkinson LZ, Ogawa Y, et al. (April 2018). "Comparative efficacy and acceptability of 21 antidepressant drugs for the acute treatment of adults with major depressive disorder: a systematic review and network meta-analysis". Lancet. 391 (10128): 1357–1366. doi:10.1016/S0140-6736(17)32802-7. PMC 5889788. PMID 29477251.
- ^ Jump up to: a b c d e f g Robinson DS, Amsterdam JD (January 2008). "The selegiline transdermal system in major depressive disorder: a systematic review of safety and tolerability". J Affect Disord. 105 (1–3): 15–23. doi:10.1016/j.jad.2007.04.024. PMID 17568687.
- ^ Jump up to: a b c d e f g h i j k l m n o p q r s t u v w x y z aa ab ac ad ae af ag ah ai Gerlach M, Youdim MB, Riederer P (December 1996). "Pharmacology of selegiline". Neurology. 47 (6 Suppl 3): S137–S145. doi:10.1212/wnl.47.6_suppl_3.137s. PMID 8959982.
- ^ Jump up to: a b c Finberg JP, Rabey JM (2016). "Inhibitors of MAO-A and MAO-B in Psychiatry and Neurology". Front Pharmacol. 7: 340. doi:10.3389/fphar.2016.00340. PMC 5067815. PMID 27803666.
- ^ Jump up to: a b c d e f g Fabbrini G, Abbruzzese G, Marconi S, Zappia M (2012). "Selegiline: a reappraisal of its role in Parkinson disease". Clin Neuropharmacol. 35 (3): 134–140. doi:10.1097/WNF.0b013e318255838b. PMID 22592509.
- ^ Jump up to: a b c d e f g h i j k l m n o Yasar S, Goldberg JP, Goldberg SR (January 1, 1996). "Are metabolites of l-deprenyl (Selegiline) useful or harmful? Indications from preclinical research". Deprenyl — Past and Future. Journal of Neural Transmission. Supplementum. Vol. 48. pp. 61–73. doi:10.1007/978-3-7091-7494-4_6. ISBN 978-3-211-82891-5. PMID 8988462.
- ^ Jump up to: a b c d Nickel B, Szelenyi I, Schulze G (December 1994). "Evaluation of physical dependence liability of l-deprenyl (selegiline) in animals". Clin Pharmacol Ther. 56 (6 Pt 2): 757–767. doi:10.1038/clpt.1994.206. PMID 7995018.
- ^ Jump up to: a b c d e f g h i j k l m n o p q r s t u v w x y z aa ab ac ad ae af ag ah ai aj ak al am an ao ap aq ar as at au av aw ax Heinonen EH, Lammintausta R (1991). "A review of the pharmacology of selegiline". Acta Neurologica Scandinavica. Supplementum. 136: 44–59. doi:10.1111/j.1600-0404.1991.tb05020.x. PMID 1686954.
- ^ Jump up to: a b c d e Knoll J (1997). "Istoriia deprenil--pervogo selektivnogo ingibitora monoaminoksidazy tipa B" [History of deprenyl--the first selective inhibitor of monoamine oxidase type B]. Voprosy Meditsinskoi Khimii. 43 (6): 482–493. PMID 9503565.
- ^ Jump up to: a b c d e f g h i j k l m n o p q r s t u Knoll J (February 1998). "(-)Deprenyl (selegiline), a catecholaminergic activity enhancer (CAE) substance acting in the brain". Pharmacol Toxicol. 82 (2): 57–66. doi:10.1111/j.1600-0773.1998.tb01399.x. PMID 9498233.
- ^ Jump up to: a b c d e f g h Miklya I (March 13, 2014). "The History of Selegiline/(-)-Deprenyl the First Selective Inhibitor of B-Type Monoamine Oxidase and The First Synthetic Catecholaminergic Activity Enhancer Substance". International Network for the History of Neuropsychopharmacology. Archived from the original on February 7, 2016. Retrieved January 7, 2016.
- ^ Jump up to: a b c d e f Gaszner P, Miklya I (January 2006). "Major depression and the synthetic enhancer substances, (-)-deprenyl and R-(-)-1-(benzofuran-2-yl)-2-propylaminopentane". Prog Neuropsychopharmacol Biol Psychiatry. 30 (1): 5–14. doi:10.1016/j.pnpbp.2005.06.004. PMID 16023777.
- ^ Jump up to: a b c d e f g h i j k l m Harsing LG, Timar J, Miklya I (August 2023). "Striking Neurochemical and Behavioral Differences in the Mode of Action of Selegiline and Rasagiline". Int J Mol Sci. 24 (17): 13334. doi:10.3390/ijms241713334. PMC 10487936. PMID 37686140.
- ^ Jump up to: a b c d e f g h i Shimazu S, Miklya I (May 2004). "Pharmacological studies with endogenous enhancer substances: β-phenylethylamine, tryptamine, and their synthetic derivatives". Prog Neuropsychopharmacol Biol Psychiatry. 28 (3): 421–427. doi:10.1016/j.pnpbp.2003.11.016. PMID 15093948.
- ^ Jump up to: a b c Berry MD (January 2007). "The potential of trace amines and their receptors for treating neurological and psychiatric diseases". Rev Recent Clin Trials. 2 (1): 3–19. doi:10.2174/157488707779318107. PMID 18473983.
- ^ Jump up to: a b c d e f g h i j k l m n o p q r Gerlach M, Reichmann H, Riederer P (2012). "A critical review of evidence for preclinical differences between rasagiline and selegiline". Basal Ganglia. 2 (4): S9–S15. doi:10.1016/j.baga.2012.04.032.
- ^ Jump up to: a b c d e f Rothman RB, Baumann MH (October 2003). "Monoamine transporters and psychostimulant drugs". Eur J Pharmacol. 479 (1–3): 23–40. doi:10.1016/j.ejphar.2003.08.054. PMID 14612135.
- ^ Jump up to: a b c d e f g h i j k Kraemer T, Maurer HH (April 2002). "Toxicokinetics of amphetamines: metabolism and toxicokinetic data of designer drugs, amphetamine, methamphetamine, and their N-alkyl derivatives". Ther Drug Monit. 24 (2): 277–289. doi:10.1097/00007691-200204000-00009. PMID 11897973.
- ^ Jump up to: a b c d e Parnham MJ (1993). "The History of l-Deprenyl". Inhibitors of Monoamine Oxidase B: Pharmacology and Clinical Use in Neurodegenerative Disorders. Milestones in Drug Therapy. Basel: Birkhäuser Basel. pp. 237–251. doi:10.1007/978-3-0348-6348-3_12. ISBN 978-3-0348-6349-0.
- ^ Jump up to: a b c d e f g h i j k Tábi T, Vécsei L, Youdim MB, Riederer P, Szökő É (May 2020). "Selegiline: a molecule with innovative potential". J Neural Transm (Vienna). 127 (5): 831–842. doi:10.1007/s00702-019-02082-0. PMC 7242272. PMID 31562557.
- ^ Jump up to: a b c d Seaman J, Landry JT (2011). Mylan: 50 Years of Unconventional Success: Making Quality Medicine Affordable and Accessible. University Press of New England. p. 50. ISBN 978-1-61168-269-4.
- ^ Jump up to: a b c d e f g h i j k l m Hoffman GR, Olson MG, Schoffstall AM, Estévez RF, Van den Eynde V, Gillman PK, et al. (December 2023). "Classics in Chemical Neuroscience: Selegiline, Isocarboxazid, Phenelzine, and Tranylcypromine". ACS Chem Neurosci. 14 (23): 4064–4075. doi:10.1021/acschemneuro.3c00591. PMID 37966854.
- ^ Jump up to: a b Golbe LI (October 1988). "Deprenyl as symptomatic therapy in Parkinson's disease". Clin Neuropharmacol. 11 (5): 387–400. doi:10.1097/00002826-198810000-00001. PMID 3146432.
- ^ Jump up to: a b c Schneider LS, Tariot PN, Goldstein B (December 1994). "Therapy with l-deprenyl (selegiline) and relation to abuse liability". Clin Pharmacol Ther. 56 (6 Pt 2): 750–756. doi:10.1038/clpt.1994.205. PMID 7995017.
- ^ Jump up to: a b c d Schifano F, Catalani V, Sharif S, Napoletano F, Corkery JM, Arillotta D, et al. (April 2022). "Benefits and Harms of 'Smart Drugs' (Nootropics) in Healthy Individuals". Drugs. 82 (6): 633–647. doi:10.1007/s40265-022-01701-7. PMID 35366192.
- ^ Blazer DG, Yaffe K, Liverman CT (July 21, 2015). Risk and Protective Factors and Interventions: General Cognitive Aging Interventions and Next Steps. National Academies Press (US). Retrieved July 5, 2024.
- ^ Jump up to: a b Brown RP, Gerbarg PL (2008). Muskin PR (ed.). "Integrative Psychopharmacology: A Practical Approach to Herbs and Nutrients in Psychiatry". Review of Psychiatry. Complementary and Alternative Medicine and Psychiatry. 19 (1). American Psychiatric Publishing: 1–66 (39). ISBN 978-1-58562-827-8. Retrieved July 5, 2024.
- ^ Jump up to: a b c d e f g h i Finberg JP (April 2019). "Inhibitors of MAO-B and COMT: their effects on brain dopamine levels and uses in Parkinson's disease". Journal of Neural Transmission. 126 (4): 433–448. doi:10.1007/s00702-018-1952-7. PMID 30386930.
- ^ Jump up to: a b c d "Drugs@FDA: FDA-Approved Drugs". accessdata.fda.gov. Retrieved July 1, 2024.
- ^ Jump up to: a b c d e f g h i j k l m Asnis GM, Henderson MA (2014). "EMSAM (deprenyl patch): how a promising antidepressant was underutilized". Neuropsychiatr Dis Treat. 10: 1911–1923. doi:10.2147/NDT.S59107. PMC 4200016. PMID 25336957.
- ^ Riederer P, Lachenmayer L, Laux G (August 2004). "Clinical applications of MAO-inhibitors". Current Medicinal Chemistry. 11 (15): 2033–2043. doi:10.2174/0929867043364775 (inactive April 2, 2024). PMID 15279566.
{{cite journal}}: CS1 maint: DOI inactive as of April 2024 (link) - ^ Jump up to: a b c d "Selegiline Hydrochloride Monograph for Professionals". Drugs.com. Retrieved February 23, 2018.
- ^ Jump up to: a b Ives NJ, Stowe RL, Marro J, Counsell C, Macleod A, Clarke CE, et al. (September 2004). "Monoamine oxidase type B inhibitors in early Parkinson's disease: meta-analysis of 17 randomised trials involving 3525 patients". BMJ. 329 (7466): 593. doi:10.1136/bmj.38184.606169.AE. PMC 516655. PMID 15310558.
- ^ Riederer P, Lachenmayer L (November 2003). "Selegiline's neuroprotective capacity revisited". Journal of Neural Transmission. 110 (11): 1273–1278. doi:10.1007/s00702-003-0083-x. PMID 14628191. S2CID 20232921.
- ^ Frisina PG, Tenenbaum HR, Borod JC, Foldi NS (May 2008). "The effects of antidepressants in Parkinson's disease: a meta-analysis". Int J Neurosci. 118 (5): 667–682. doi:10.1080/00207450701239418. PMID 18446583.
- ^ Tsuboi T, Satake Y, Hiraga K, Yokoi K, Hattori M, Suzuki M, et al. (June 2022). "Effects of MAO-B inhibitors on non-motor symptoms and quality of life in Parkinson's disease: A systematic review". npj Parkinsons Dis. 8 (1): 75. doi:10.1038/s41531-022-00339-2. PMC 9192747. PMID 35697709.
- ^ Jump up to: a b c Binde CD, Tvete IF, Gåsemyr J, Natvig B, Klemp M (September 2018). "A multiple treatment comparison meta-analysis of monoamine oxidase type B inhibitors for Parkinson's disease". Br J Clin Pharmacol. 84 (9): 1917–1927. doi:10.1111/bcp.13651. PMC 6089809. PMID 29847694.
- ^ Hengartner MP, Jakobsen JC, Sørensen A, Plöderl M (2020). "Efficacy of new-generation antidepressants assessed with the Montgomery-Asberg Depression Rating Scale, the gold standard clinician rating scale: A meta-analysis of randomised placebo-controlled trials". PLOS ONE. 15 (2): e0229381. Bibcode:2020PLoSO..1529381H. doi:10.1371/journal.pone.0229381. PMC 7043778. PMID 32101579.
- ^ Jump up to: a b c d e f g h i j k l m n o p q r Cristancho MA, Thase ME (2016). "Critical appraisal of selegiline transdermal system for major depressive disorder". Expert Opinion on Drug Delivery. 13 (5): 659–665. doi:10.1517/17425247.2016.1140145. PMID 26837935.
- ^ Pae CU, Patkar AA, Jang S, Portland KB, Jung S, Nelson JC (August 2014). "Efficacy and safety of selegiline transdermal system (STS) for the atypical subtype of major depressive disorder: pooled analysis of 5 short-term, placebo-controlled trials". CNS Spectr. 19 (4): 324–329. doi:10.1017/S1092852913000655. PMID 24168807.
- ^ Jump up to: a b c d e Clayton AH, Campbell BJ, Favit A, Yang Y, Moonsammy G, Piontek CM, et al. (December 2007). "Symptoms of sexual dysfunction in patients treated for major depressive disorder: a meta-analysis comparing selegiline transdermal system and placebo using a patient-rated scale". J Clin Psychiatry. 68 (12): 1860–1866. doi:10.4088/jcp.v68n1205. PMID 18162016.
- ^ Winter J, Curtis K, Hu B, Clayton AH (July 2022). "Sexual dysfunction with major depressive disorder and antidepressant treatments: impact, assessment, and management". Expert Opin Drug Saf. 21 (7): 913–930. doi:10.1080/14740338.2022.2049753. PMID 35255754.
- ^ Jump up to: a b c d e f g h i j Knoll J (1983). "Deprenyl (selegiline): the history of its development and pharmacological action". Acta Neurol Scand Suppl. 95: 57–80. doi:10.1111/j.1600-0404.1983.tb01517.x. PMID 6428148.
- ^ Jump up to: a b c d e f Alborghetti M, Nicoletti F (2019). "Different Generations of Type-B Monoamine Oxidase Inhibitors in Parkinson's Disease: From Bench to Bedside". Curr Neuropharmacol. 17 (9): 861–873. doi:10.2174/1570159X16666180830100754. PMC 7052841. PMID 30160213.
- ^ Friedman RA, Leon AC (June 2007). "Expanding the black box - depression, antidepressants, and the risk of suicide". The New England Journal of Medicine. 356 (23): 2343–2346. doi:10.1056/NEJMp078015. PMID 17485726.
- ^ Jump up to: a b c d Patkar AA, Pae CU, Masand PS (May 2006). "Transdermal selegiline: the new generation of monoamine oxidase inhibitors". CNS Spectr. 11 (5): 363–375. doi:10.1017/s1092852900014498. PMID 16641841.
- ^ Jump up to: a b Finberg JP, Gillman K (2011). Selective inhibitors of monoamine oxidase type B and the "cheese effect". International Review of Neurobiology. Vol. 100. pp. 169–190. doi:10.1016/B978-0-12-386467-3.00009-1. ISBN 978-0-12-386467-3. PMID 21971008.
- ^ Olanow CW, Myllylä VV, Sotaniemi KA, Larsen JP, Pålhagen S, Przuntek H, et al. (September 1998). "Effect of selegiline on mortality in patients with Parkinson's disease: a meta-analysis". Neurology. 51 (3): 825–830. doi:10.1212/wnl.51.3.825. PMID 9748034.
- ^ Aaltonen H, Kilkku O, Heinonen E, Mäki-Ikola O (December 1998). "Effect of adding selegiline to levodopa in early, mild Parkinson's disease. Evidence is insufficient to show that combined treatment increases mortality". BMJ. 317 (7172): 1586–1587. doi:10.1136/bmj.317.7172.1586. PMC 1114394. PMID 9890764.
- ^ Abassi ZA, Binah O, Youdim MB (October 2004). "Cardiovascular activity of rasagiline, a selective and potent inhibitor of mitochondrial monoamine oxidase B: comparison with selegiline". Br J Pharmacol. 143 (3): 371–378. doi:10.1038/sj.bjp.0705962. PMC 1575354. PMID 15339864.
- ^ Jump up to: a b Roy MA, Doiron M, Talon-Croteau J, Dupré N, Simard M (July 2018). "Effects of Antiparkinson Medication on Cognition in Parkinson's Disease: A Systematic Review". Can J Neurol Sci. 45 (4): 375–404. doi:10.1017/cjn.2018.21. PMID 29747716.
- ^ Jump up to: a b Vitale C, Amboni M, Erro R, Picillo M, Pellecchia MT, Barone P, et al. (June 2019). "Parkinson's disease management and impulse control disorders: current state and future perspectives". Expert Rev Neurother. 19 (6): 495–508. doi:10.1080/14737175.2019.1620603. PMID 31148487.
- ^ Jump up to: a b Djamshidian A, Cardoso F, Grosset D, Bowden-Jones H, Lees AJ (September 2011). "Pathological gambling in Parkinson's disease--a review of the literature". Mov Disord. 26 (11): 1976–1984. doi:10.1002/mds.23821. PMID 21661054.
- ^ Drapier D, Drapier S, Sauleau P, Derkinderen P, Damier P, Allain H, et al. (November 2006). "Pathological gambling secondary to dopaminergic therapy in Parkinson's disease". Psychiatry Res. 144 (2–3): 241–244. doi:10.1016/j.psychres.2006.04.017. PMID 17011634.
- ^ Solla P, Bortolato M, Cannas A, Mulas CS, Marrosu F (April 2015). "Paraphilias and paraphilic disorders in Parkinson's disease: A systematic review of the literature". Mov Disord. 30 (5): 604–613. doi:10.1002/mds.26157. PMC 4428164. PMID 25759330.
- ^ Hirao K, Kaneko Y, Hirose D, Fukasawa R, Shimizu S, Kanetaka H, et al. (September 2019). "Patient with Parkinson's disease presenting with impulse control disorders following treatment with selegiline". Int Psychogeriatr. 31 (9): 1375–1376. doi:10.1017/S1041610218001862. PMID 30520410.
- ^ Uitti RJ, Tanner CM, Rajput AH, Goetz CG, Klawans HL, Thiessen B (October 1989). "Hypersexuality with antiparkinsonian therapy". Clin Neuropharmacol. 12 (5): 375–383. doi:10.1097/00002826-198910000-00002. PMID 2575449.
- ^ Riley DE (2002). "Reversible transvestic fetishism in a man with Parkinson's disease treated with selegiline". Clin Neuropharmacol. 25 (4): 234–237. doi:10.1097/00002826-200207000-00008. PMID 12151912.
- ^ Shapiro MA, Chang YL, Munson SK, Okun MS, Fernandez HH (September 2006). "Hypersexuality and paraphilia induced by selegiline in Parkinson's disease: report of 2 cases". Parkinsonism Relat Disord. 12 (6): 392–395. doi:10.1016/j.parkreldis.2006.01.010. PMID 16730214.
- ^ Howell M, Avidan AY, Foldvary-Schaefer N, Malkani RG, During EH, Roland JP, et al. (April 2023). "Management of REM sleep behavior disorder: an American Academy of Sleep Medicine clinical practice guideline". J Clin Sleep Med. 19 (4): 759–768. doi:10.5664/jcsm.10424. PMC 10071384. PMID 36515157.
- ^ Hoque R, Chesson AL (February 2010). "Pharmacologically induced/exacerbated restless legs syndrome, periodic limb movements of sleep, and REM behavior disorder/REM sleep without atonia: literature review, qualitative scoring, and comparative analysis". J Clin Sleep Med. 6 (1): 79–83. doi:10.5664/jcsm.27716. PMC 2823282. PMID 20191944.
- ^ Louden MB, Morehead MA, Schmidt HS (1995). "Activation by selegiline (Eldepryle) of REM sleep behavior disorder in parkinsonism". W V Med J. 91 (3): 101. PMID 7747490.
- ^ Jump up to: a b c Goldberg SR, Yasar S, Bergman J, Youdim MB (December 1994). "Introduction: examination of clinical and preclinical pharmacologic data relating to abuse liability of l-deprenyl (selegiline)". Clin Pharmacol Ther. 56 (6 Pt 2): 721–724. doi:10.1038/clpt.1994.201. PMID 7995013.
- ^ Jump up to: a b Winger GD, Yasar S, Negus SS, Goldberg SR (December 1994). "Intravenous self-administration studies with l-deprenyl (selegiline) in monkeys". Clin Pharmacol Ther. 56 (6 Pt 2): 774–780. doi:10.1038/clpt.1994.208. hdl:2027.42/110034. PMID 7995020.
- ^ Jump up to: a b Yasar S, Gaál J, Panlilio LV, Justinova Z, Molnár SV, Redhi GH, et al. (January 2006). "A comparison of drug-seeking behavior maintained by D-amphetamine, L-deprenyl (selegiline), and D-deprenyl under a second-order schedule in squirrel monkeys". Psychopharmacology (Berl). 183 (4): 413–421. doi:10.1007/s00213-005-0200-7. PMC 1360227. PMID 16292593.
- ^ McKean AJ, Leung JG, Dare FY, Sola CL, Schak KM (2015). "The Perils of Illegitimate Online Pharmacies: Substance-Induced Panic Attacks and Mood Instability Associated With Selegiline and Phenylethylamine". Psychosomatics. 56 (5): 583–587. doi:10.1016/j.psym.2015.05.003. PMID 26198572.
- ^ Monteith S, Glenn T, Bauer R, Conell J, Bauer M (March 2016). "Availability of prescription drugs for bipolar disorder at online pharmacies". J Affect Disord. 193: 59–65. doi:10.1016/j.jad.2015.12.043. PMID 26766033.
- ^ Kuhn W, Müller T (1996). "The clinical potential of Deprenyl in neurologic and psychiatric disorders". Deprenyl — Past and Future. Vol. 48. pp. 85–93. doi:10.1007/978-3-7091-7494-4_8. ISBN 978-3-211-82891-5. PMID 8988464.
{{cite book}}:|journal=ignored (help) - ^ Jump up to: a b c Heinonen EH, Myllylä V (July 1998). "Safety of selegiline (deprenyl) in the treatment of Parkinson's disease". Drug Safety. 19 (1): 11–22. doi:10.2165/00002018-199819010-00002. PMID 9673855. S2CID 9632549.
- ^ Csoti I, Storch A, Müller W, Jost WH (December 1, 2012). "Drug interactions with selegiline versus rasagiline". Basal Ganglia. Monoamine oxidase B Inhibitors. 2 (4, Supplement): S27–S31. doi:10.1016/j.baga.2012.06.003. ISSN 2210-5336.
- ^ Gillman PK (October 2005). "Monoamine oxidase inhibitors, opioid analgesics and serotonin toxicity". British Journal of Anaesthesia. 95 (4): 434–441. doi:10.1093/bja/aei210. PMID 16051647.
- ^ Jessen L, Kovalick LJ, Azzaro AJ (April 2008). "The selegiline transdermal system (emsam): a therapeutic option for the treatment of major depressive disorder". P & T. 33 (4): 212–246. PMC 2730099. PMID 19750165.
- ^ Azzaro AJ, VanDenBerg CM, Ziemniak J, Kemper EM, Blob LF, Campbell BJ (August 2007). "Evaluation of the potential for pharmacodynamic and pharmacokinetic drug interactions between selegiline transdermal system and two sympathomimetic agents (pseudoephedrine and phenylpropanolamine) in healthy volunteers". J Clin Pharmacol. 47 (8): 978–90. doi:10.1177/0091270007302950. PMID 17554106.
- ^ Jump up to: a b Elkashef A, Vocci F, Hanson G, White J, Wickes W, Tiihonen J (2008). "Pharmacotherapy of methamphetamine addiction: an update". Subst Abus. 29 (3): 31–49. doi:10.1080/08897070802218554. PMC 2597382. PMID 19042205.
- ^ Jump up to: a b Newton TF, De La Garza R, Fong T, Chiang N, Holmes TH, Bloch DA, et al. (December 2005). "A comprehensive assessment of the safety of intravenous methamphetamine administration during treatment with selegiline". Pharmacol Biochem Behav. 82 (4): 704–711. doi:10.1016/j.pbb.2005.11.012. PMID 16413604.
- ^ Jump up to: a b Finberg JP (August 2014). "Update on the pharmacology of selective inhibitors of MAO-A and MAO-B: focus on modulation of CNS monoamine neurotransmitter release". Pharmacol Ther. 143 (2): 133–152. doi:10.1016/j.pharmthera.2014.02.010. PMID 24607445.
- ^ Jump up to: a b Houtsmuller EJ, Notes LD, Newton T, van Sluis N, Chiang N, Elkashef A, et al. (February 2004). "Transdermal selegiline and intravenous cocaine: safety and interactions". Psychopharmacology (Berl). 172 (1): 31–40. doi:10.1007/s00213-003-1616-6. PMID 14605792.
- ^ Jump up to: a b Bartzokis G, Beckson M, Newton T, Mandelkern M, Mintz J, Foster JA, et al. (June 1999). "Selegiline effects on cocaine-induced changes in medial temporal lobe metabolism and subjective ratings of euphoria". Neuropsychopharmacology. 20 (6): 582–590. doi:10.1016/S0893-133X(98)00092-X. PMID 10327427.
- ^ Jump up to: a b Haberny KA, Walsh SL, Ginn DH, Wilkins JN, Garner JE, Setoda D, et al. (July 1995). "Absence of acute cocaine interactions with the MAO-B inhibitor selegiline". Drug Alcohol Depend. 39 (1): 55–62. doi:10.1016/0376-8716(95)01137-n. PMID 7587975.
- ^ Jump up to: a b Harris DS, Everhart T, Jacob P, Lin E, Mendelson JE, Jones RT (August 2009). "A phase 1 trial of pharmacologic interactions between transdermal selegiline and a 4-hour cocaine infusion". BMC Clin Pharmacol. 9: 13. doi:10.1186/1472-6904-9-13. PMC 2731040. PMID 19646280.
- ^ Jump up to: a b Newton TF, Kalechstein A, Beckson M, Bartzokis G, Bridge TP, Ling W (October 1999). "Effects of selegiline pretreatment on response to experimental cocaine administration". Psychiatry Res. 87 (2–3): 101–106. doi:10.1016/s0165-1781(99)00058-x. PMID 10579543.
- ^ Feinberg SS (November 2004). "Combining stimulants with monoamine oxidase inhibitors: a review of uses and one possible additional indication". J Clin Psychiatry. 65 (11): 1520–1524. doi:10.4088/jcp.v65n1113. PMID 15554766.
- ^ Thomas SJ, Shin M, McInnis MG, Bostwick JR (April 2015). "Combination therapy with monoamine oxidase inhibitors and other antidepressants or stimulants: strategies for the management of treatment-resistant depression". Pharmacotherapy. 35 (4): 433–449. doi:10.1002/phar.1576. hdl:2027.42/111275. PMID 25884531.
- ^ Israel JA (2015). "Combining Stimulants and Monoamine Oxidase Inhibitors: A Reexamination of the Literature and a Report of a New Treatment Combination". Prim Care Companion CNS Disord. 17 (6). doi:10.4088/PCC.15br01836. PMC 4805402. PMID 27057401.
- ^ Culpepper L, Kovalick LJ (2008). "A review of the literature on the selegiline transdermal system: an effective and well-tolerated monoamine oxidase inhibitor for the treatment of depression". Prim Care Companion J Clin Psychiatry. 10 (1): 25–30. doi:10.4088/pcc.v10n0105. PMC 2249821. PMID 18311418.
- ^ Eccles R (January 2007). "Substitution of phenylephrine for pseudoephedrine as a nasal decongeststant. An illogical way to control methamphetamine abuse". Br J Clin Pharmacol. 63 (1): 10–14. doi:10.1111/j.1365-2125.2006.02833.x. PMC 2000711. PMID 17116124.
- ^ Richards E, Lopez MJ, Maani CV (2023). "Phenylephrine". StatPearls. Treasure Island, Florida: StatPearls Publishing. PMID 30521222. Retrieved April 27, 2023.
- ^ Jump up to: a b c d Гиллман П.К. (февраль 2011 г.). «Достижения в области фармакологии и взаимодействия необратимых неселективных ингибиторов моноаминоксидазы». Дж. Клин Психофармакол . 31 (1): 66–74. doi : 10.1097/JCP.0b013e31820469ea . ПМИД 21192146 .
- ^ Jump up to: а б с д Гиллман П.К. (ноябрь 2018 г.). «Переоценка профиля безопасности ингибиторов моноаминоксидазы: разъяснение старых устаревших мифов о тираминах». J Neural Transm (Вена) . 125 (11): 1707–1717. дои : 10.1007/s00702-018-1932-y . ПМИД 30255284 .
- ^ Jump up to: а б с д Ван ден Эйнде В., Годе Л., Редхед С., Хорвиц А., Барнетт Б. (2023). «Ингибиторы моноаминоксидазы и клинически значимые лекарственные взаимодействия: руководство по предотвращению серотониновой токсичности и гипертензивных реакций». Психиатрические летописи . 53 (8): 353–358. дои : 10.3928/00485713-20230713-02 . ISSN 0048-5713 .
- ^ Jump up to: а б Ван ден Эйнде В., Абдельмоемин В.Р., Авраам М.М., Амстердам Дж.Д., Андерсон И.М., Андраде С. и др. (июль 2022 г.). «Руководство для врача по классическим ингибиторам МАО (фенелзин, транилципромин, изокарбоксазид) при резистентной к лечению депрессии». Спектр ЦНС . 28 (4): 427–440. дои : 10.1017/S1092852922000906 . hdl : 2292/61637 . ПМИД 35837681 .
- ^ Jump up to: а б с д и ж Шейнин Х., Анттила М., Даль М.Л., Карнани Х., Найман Л., Таавитсайнен П. и др. (октябрь 1998 г.). «Полиморфизм CYP2D6 не имеет решающего значения для распределения селегилина». Клин Фармакол Тер . 64 (4): 402–411. дои : 10.1016/S0009-9236(98)90071-6 . ПМИД 9797797 .
- ^ Jump up to: а б с д и Лайне К., Анттила М., Найман Л., Уолберг А., Бертилссон Л. (май 2001 г.). «Полиморфизм CYP2C19 не важен для метаболизма селегилина in vivo». Эур Дж Клин Фармакол . 57 (2): 137–142. дои : 10.1007/s002280100289 . ПМИД 11417445 .
- ^ Jump up to: а б Кивистё К.Т., Ван Дж.С., Бэкман Дж.Т., Найман Л., Таавитсайнен П., Анттила М. и др. (апрель 2001 г.). «Ингибитор CYP3A4 итраконазол не влияет на фармакокинетику селегилина». Эур Дж Клин Фармакол . 57 (1): 37–42. дои : 10.1007/s002280100278 . ПМИД 11372588 .
- ^ Jump up to: а б «Таблица субстратов, ингибиторов и индукторов» . Управление по контролю за продуктами и лекарствами США . 5 июня 2023 г. . Проверено 5 июля 2024 г.
- ^ Наой М., Маруяма В., Шамото-Нагай М. (сентябрь 2022 г.). «Нейропротекторная функция разагилина и селегилина, ингибиторов моноаминоксидазы типа B и роль моноаминоксидазы в синуклеинопатиях» . Int J Mol Sci . 23 (19): 11059. doi : 10.3390/ijms231911059 . ПМЦ 9570229 . ПМИД 36232361 .
- ^ Jump up to: а б с д Клитц М., Гретен С., Вегнер Ф., Хёглингер Г.Ю. (июнь 2019 г.). «Безопасность и переносимость фармакотерапии болезни Паркинсона у гериатрических пациентов». Наркотики и старение . 36 (6): 511–530. дои : 10.1007/s40266-019-00654-z . ПМИД 30937878 .
- ^ Jump up to: а б с д Лайне К., Анттила М., Хелминен А., Карнани Х., Хууппонен Р. (март 1999 г.). «Исследование линейности дозы фармакокинетики селегилина после перорального приема: доказательства сильного взаимодействия препарата с женскими половыми стероидами» . Бр Дж Клин Фармакол . 47 (3): 249–254. дои : 10.1046/j.1365-2125.1999.00891.x . ПМК 2014223 . ПМИД 10215747 .
- ^ Jump up to: а б Паловаара С., Анттила М., Найман Л., Лэйн К. (июль 2002 г.). «Влияние сопутствующей заместительной гормональной терапии, содержащей эстрадиол и левоноргестрел, на фармакокинетику селегилина». Европейский журнал клинической фармакологии . 58 (4): 259–263. дои : 10.1007/s00228-002-0469-y . ПМИД 12136372 .
- ^ Jump up to: а б с д и ж г час я Анттила М., Сотаниеми Э.А., Пелконен О., Раутио А. (январь 2005 г.). «Выраженное влияние функции печени и почек на фармакокинетику селегилина». Клин Фармакол Тер . 77 (1): 54–62. дои : 10.1016/j.clpt.2004.09.004 . ПМИД 15637531 .
- ^ Jump up to: а б с д и Зангер У.М., Кляйн К. (2013). «Фармакогенетика цитохрома P450 2B6 (CYP2B6): достижения в области полиморфизма, механизмов и клинической значимости» . Фронт Генет . 4 : 24. дои : 10.3389/fgene.2013.00024 . ПМЦ 3588594 . ПМИД 23467454 .
- ^ Jump up to: а б с Хедрих В.Д., Хасан Х.Э., Ван Х. (сентябрь 2016 г.). «Информация о лекарственном взаимодействии, опосредованном CYP2B6» . Акта Фарм Син Б. 6 (5): 413–425. дои : 10.1016/j.apsb.2016.07.016 . ПМК 5045548 . ПМИД 27709010 .
- ^ Jump up to: а б с д Шридар С., Кенаан С., Холленберг П.Ф. (декабрь 2012 г.). «Ингибирование метаболизма бупропиона селегилином: механизм инактивации человеческого CYP2B6 и характеристика глутатиона и пептидных аддуктов» . Препарат Метаб Диспос . 40 (12): 2256–2266. дои : 10.1124/dmd.112.046979 . ПМК 3500550 . ПМИД 22936314 .
- ^ Jump up to: а б Нироги Р., Палачарла Р.К., Мохаммед А.Р., Манохаран А., Поннаманени Р.К., Бхирапунени Г. (март 2015 г.). «Оценка метаболизмзависимого ингибирования гидроксилирования бупропиона, опосредованного CYP2B6, в микросомах печени человека ингибиторами моноаминоксидазы и прогнозирование потенциальных виновников лекарственного взаимодействия». Хим-биол-взаимодействие . 230 : 9–20. Бибкод : 2015CBI...230....9N . дои : 10.1016/j.cbi.2015.01.028 . ПМИД 25656918 .
- ^ Риттер Дж.Л., Александр Б. (март 1997 г.). «Ретроспективное исследование лекарственного взаимодействия селегилина с антидепрессантами и обзор литературы». Энн Клин Психиатрия . 9 (1): 7–13. дои : 10.1023/а:1026222106851 . ПМИД 9167831 .
- ^ Таннер Дж. А., Тиндейл РФ (декабрь 2017 г.). «Вариации активности CYP2A6 и персонализированная медицина» . Дж. Перс Мед . 7 (4): 18. дои : 10.3390/jpm7040018 . ПМЦ 5748630 . ПМИД 29194389 .
- ^ Jump up to: а б с д и Сиу ЕС, Тиндейл РФ (март 2008 г.). «Селегилин представляет собой инактиватор CYP2A6, ингибирующий метаболизм никотина у людей и мышей». J Pharmacol Exp Ther . 324 (3): 992–9. дои : 10.1124/jpet.107.133900 . ПМИД 18065502 .
- ^ Jump up to: а б с Лайне К., Анттила М., Хууппонен Р., Мяки-Икола О., Хейнонен Э. (2000). «Фармакокинетика многократных доз селегилина и десметилселегилина предполагает насыщаемое связывание с тканями». Клин Нейрофармакол . 23 (1): 22–27. дои : 10.1097/00002826-200001000-00005 . ПМИД 10682227 .
- ^ Пфайффер РФ (май 1996 г.). «Противопаркинсонические средства. Лекарственные взаимодействия, имеющие клиническое значение». Препарат Саф . 14 (5): 343–354. дои : 10.2165/00002018-199614050-00006 . ПМИД 8800629 .
- ^ Нолл Дж. (сентябрь 1992 г.). «Фармакологические основы терапевтического действия (-)депренила при возрастных неврологических заболеваниях». Медресе преп . 12 (5): 505–524. дои : 10.1002/мед.2610120504 . ПМИД 1513186 .
- ^ Jump up to: а б с д и ж г Нолл Дж. (май 1992 г.). «Фармакологический профиль (-) депренила (селегилина) и его актуальность для человека: личный взгляд». Фармакология и токсикология . 70 (5, часть 1): 317–321. дои : 10.1111/j.1600-0773.1992.tb00480.x . ПМИД 1608919 .
- ^ Jump up to: а б Биркс Дж., Фликер Л. (2003). «Селегилин от болезни Альцгеймера». Cochrane Database Syst Rev (1): CD000442. дои : 10.1002/14651858.CD000442 . ПМИД 12535396 .
- ^ Мадьяр К., Палфи М., Таби Т., Калас Х., Сзенде Б., Сёко Э. (август 2004 г.). «Фармакологические аспекты (-)-депренила». Curr Med Chem . 11 (15): 2017–31. дои : 10.2174/0929867043364793 . ПМИД 15279565 .
- ^ Мадьяр К., Сзенде Б (январь 2004 г.). «(-)-Депренил, селективный ингибитор МАО-В с апоптотическим и антиапоптотическим свойствами». Нейротоксикология . 25 (1–2): 233–242. Бибкод : 2004NeuTx..25..233M . дои : 10.1016/S0161-813X(03)00102-5 . ПМИД 14697898 .
- ^ Jump up to: а б Наой М., Маруяма В., Шамото-Нагай М., Ридерер П. (июнь 2024 г.). «Токсические взаимодействия между дофамином, α-синуклеином, моноаминоксидазой и генами в митохондриях болезни Паркинсона». J Neural Transm (Вена) . 131 (6): 639–661. дои : 10.1007/s00702-023-02730-6 . ПМИД 38196001 .
- ^ Ореланд Л., Йоханссон Ф., Экстедт Дж. (1983). «Режим дозирования депренила (селегилина) и активности МАО тромбоцитов». Acta Neurologica Scandinavica. Дополнение . 95 : 87–89. дои : 10.1111/j.1600-0404.1983.tb01519.x . ПМИД 6428150 .
- ^ Jump up to: а б с Махмуд I (декабрь 1998 г.). «Являются ли 10 миллиграммов селегилина необходимыми в качестве вспомогательной терапии для симптоматического лечения болезни Паркинсона?». The Drug Monit . 20 (6): 717–721. дои : 10.1097/00007691-199812000-00024 . ПМИД 9853994 .
- ^ Мюллер Т. (октябрь 2014 г.). «Фармакокинетическая/фармакодинамическая оценка мезилата разагилина при болезни Паркинсона». Экспертное мнение о препарате Метаб Токсикол . 10 (10): 1423–32. дои : 10.1517/17425255.2014.943182 . ПМИД 25196265 .
- ^ Багхай Т.К., Эсер Д., Шуле С., Борн С., Рупрехт Р. (25 октября 2007 г.). «Селегилиновая трансдермальная система в лечении депрессивных расстройств». Будущая неврология . 2 (6): 601–611. дои : 10.2217/14796708.2.6.601 . ISSN 1479-6708 .
- ^ Тейченн П.Ф., Паркер С. (1989). «Двойное слепое перекрестное плацебо-контролируемое исследование селегилина при болезни Паркинсона - промежуточный анализ». Acta Neurologica Scandinavica. Дополнение . 126 : 119–125. дои : 10.1111/j.1600-0404.1989.tb01791.x . ПМИД 2515717 .
- ^ Jump up to: а б Ридерер П., Юдим М.Б. (май 1986 г.). «Активность моноаминоксидазы и метаболизм моноаминов в мозге пациентов с паркинсонизмом, получавших l-депренил». Журнал нейрохимии . 46 (5): 1359–1365. дои : 10.1111/j.1471-4159.1986.tb01747.x . ПМИД 2420928 .
- ^ Jump up to: а б Фаулер Дж.С., Волков Н.Д., Логан Дж., Ван Г.Дж., МакГрегор Р.Р., Шайлер Д. и др. (октябрь 1994 г.). «Медленное восстановление МАО Б головного мозга человека после отмены L-депренила (селегелина)». Синапс . 18 (2): 86–93. дои : 10.1002/syn.890180203 . ПМИД 7839316 .
- ^ Jump up to: а б с Ридерер П., Йеллингер К., Зееманн Д. (1984). «Моноаминоксидаза и паркинсонизм» . Типтон К.Ф., Достерт П., Бенедетти М.С. (ред.). Моноаминоксидаза и заболевания: перспективы терапии обратимыми ингибиторами . Быстрое воспроизведение рукописей Academic Press. Академическая пресса. стр. 404–415. ISBN 978-0-12-691660-7 . Проверено 5 июля 2024 г.
- ^ Ридерер П., Юдим М.Б., Рауш В.Д., Биркмайер В., Йеллингер К., Зееманн Д. (1978). «О механизме действия L-депренила на центральную нервную систему человека». Журнал нейронной передачи . 43 (3–4): 217–226. дои : 10.1007/BF01246958 . ПМИД 745014 .
- ^ Рейнольдс Г.П., Ридерер П., Сандлер М., Джеллингер К., Зееманн Д. (1978). «Амфетамин и 2-фенилэтиламин в посмертном мозге с болезнью Паркинсона после введения (-) депренила». Журнал нейронной передачи . 43 (3–4): 271–277. дои : 10.1007/BF01246964 . ПМИД 745019 .
- ^ Jump up to: а б Фаулер Дж.С., Логан Дж., Волков Н.Д., Шумей Э., Макколл-Перес Ф., Джейн М. и др. (февраль 2015 г.). «Доказательства того, что составы селективного ингибитора МАО-В, селегилина, которые обходят метаболизм первого прохождения, также ингибируют МАО-А в мозге человека» . Нейропсихофармакология . 40 (3): 650–657. дои : 10.1038/нпп.2014.214 . ПМЦ 4289953 . ПМИД 25249059 .
- ^ Биркмайер В., Ридерер П., Юдим М.Б. (1982). «(-) Депренил в лечении болезни Паркинсона». Клин Нейрофармакол . 5 (2): 195–230. дои : 10.1097/00002826-198205020-00004 . ПМИД 6814755 .
- ^ Элсворт Дж.Д., Гловер В., Рейнольдс Г.П., Сэндлер М., Лис А.Дж., Фуапрадит П. и др. (апрель 1978 г.). «Введение депренила человеку: селективный ингибитор моноаминоксидазы B без «сырного эффекта» ». Психофармакология (Берл) . 57 (1): 33–38. дои : 10.1007/BF00426954 . ПМИД 96466 .
- ^ Jump up to: а б с Янссен П.А., Лейсен Дж.Э., Мегенс А.А., Авоутерс Ф.Х. (сентябрь 1999 г.). «Действует ли фенилэтиламин у некоторых пациентов как эндогенный амфетамин?». Int J Нейропсихофармакол . 2 (3): 229–240. дои : 10.1017/S1461145799001522 . ПМИД 11281991 .
- ^ Ридерер П., Ло Г. (2011). «Ингибиторы МАО при болезни Паркинсона» . Опыт Нейробиола . 20 (1): 1–17. дои : 10.5607/en.2011.20.1.1 . ПМЦ 3213739 . ПМИД 22110357 .
- ^ Jump up to: а б Ясар С., Юстинова З., Ли Ш., Стефански Р., Голдберг С.Р., Танда Г. (апрель 2006 г.). «Метаболическая трансформация играет первостепенную роль в психостимулирующих дискриминативно-стимулирующих эффектах селегилина [(R)-(-)-депренила]». J Pharmacol Exp Ther . 317 (1): 387–394. дои : 10.1124/jpet.105.096263 . ПМИД 16352699 .
- ^ Jump up to: а б Мадьяр К., Визи Э.С., Эчери З., Нолл Дж. (1967). «Сравнительный фармакологический анализ оптических изомеров фенил-изопропил-метилпропиниламина (Е-250)». Acta Physiologica Academiae Scientiarum Hungaricae . 32 (4): 377–387. ПМИД 5595908 .
- ^ Jump up to: а б Хейнонен Э.Х., Анттила М.И., Карнани Х.Л., Найман Л.М., Вуоринен Ю.А., Пюиккё К.А. и др. (июль 1997 г.). «Десметилселегилин, метаболит селегилина, является необратимым ингибитором моноаминоксидазы типа B у человека». Джей Клин Фармакол . 37 (7): 602–609. дои : 10.1002/j.1552-4604.1997.tb04342.x . ПМИД 9243353 .
- ^ Jump up to: а б с Надим М.С., Хосави С.Б., Муртаза Б.Н., Казми I (2023). «Механизм действия противопаркинсонических препаратов». Как работают синтетические лекарства: взгляд на молекулярную фармакологию классических и новых фармацевтических препаратов . Эльзевир. стр. 195–213. дои : 10.1016/b978-0-323-99855-0.00009-9 . ISBN 978-0-323-99855-0 .
- ^ Jump up to: а б с Нам М.Х., Са М., Джу Ю.Х., Пак М.Г., Ли СиДжей (апрель 2022 г.). «Возвращаясь к роли астроцитарного МАОБ в болезни Паркинсона» . Международный журнал молекулярных наук . 23 (8): 4453. doi : 10.3390/ijms23084453 . ПМЦ 9028367 . ПМИД 35457272 .
- ^ Jump up to: а б с Чо ХУ, Ким С., Сим Дж., Ян С., Ан Х., Нам М.Х. и др. (июль 2021 г.). «Переопределение дифференциальной роли МАО-А в деградации дофамина и МАО-В в тоническом синтезе ГАМК» . Экспериментальная и молекулярная медицина . 53 (7): 1148–1158. дои : 10.1038/s12276-021-00646-3 . ПМЦ 8333267 . ПМИД 34244591 .
- ^ Jump up to: а б с д и ж г час Нолл Дж (2005). «Регуляция усилителей: нейрохимический подход к врожденным и приобретенным побуждениям». Мозг и он сам: нейрохимическая концепция врожденных и приобретенных влечений . Берлин/Гейдельберг: Springer-Verlag. стр. 25–94. дои : 10.1007/3-540-27434-0_4 . ISBN 978-3-540-23969-7 .
- ^ Jump up to: а б с д и ж г час я дж к л м н тот п д р Нолл Дж (2001). «Антивозрастные соединения: (-)депренил (селегелин) и (-)1-(бензофуран-2-ил)-2-пропиламинопентан, [(-)BPAP], селективный высокоэффективный усилитель высвобождения катехоламинов, опосредованного распространением импульса и серотонин в мозгу» . Препарат для ЦНС Rev. 7 (3): 317–345. дои : 10.1111/j.1527-3458.2001.tb00202.x . ПМК 6494119 . ПМИД 11607046 .
- ^ Jump up to: а б с д и ж г час я Нолл Дж (2012). Как селегилин ((-)-депренил) замедляет старение мозга . Издательство Bentham Science. стр. 16, 43, 70, 86, 90, 92. ISBN. 978-1-60805-470-1 . Проверено 4 июля 2024 г.
- ^ Jump up to: а б с д и Калас Х., Мадьяр К., Секе Э, Адегате Э., Адем А., Хасан М.Ю. и др. (2014). «Метаболизм селегилина [(-)-депренила)]». Curr Med Chem . 21 (13): 1522–1530. дои : 10.2174/0929867321666131218094352 . ПМИД 24350849 .
- ^ Jump up to: а б с д Нолл Дж. (август 2003 г.). «Усилитель регуляции / эндогенные и синтетические соединения-усилители: нейрохимическая концепция врожденных и приобретенных влечений». Нейрохим Рез . 28 (8): 1275–1297. дои : 10.1023/а:1024224311289 . ПМИД 12834268 .
- ^ Нолл Дж., Микля И., Нолл Б., Ясуса Т., Симадзу С., Йонеда Ф. (сентябрь 2002 г.). «1-(Бензофуран-2-ил)-2-(3,3,3-трифторпропил)аминопентан HCl, 3-F-BPAP, противодействует усиливающему эффекту (-)-BPAP в челночном боксе и оставляет эффект (-)-депренил без изменений». Наука о жизни . 71 (17): 1975–84. дои : 10.1016/s0024-3205(02)01968-9 . ПМИД 12175892 .
- ^ Jump up to: а б с Нолл Дж., Зелена Д., Тимар Дж., Баги К., Мерваи З., Микля И. (январь 2020 г.). «Синтетические соединения-усилители, помимо воздействия на биогенную аминную систему, влияют на передачу глутамата и реакцию на стресс». Поведение мозга Res . 378 : 112290. doi : 10.1016/j.bbr.2019.112290 . ПМИД 31610214 .
- ^ Симадзу С., Цунэкава Х., Йонеда Ф., Кацуки Х., Акаике А., Яновский А. (декабрь 2003 г.). «Опосредованное транспортером действие R-(-)-1-(бензофуран-2-ил)-2-пропиламинопентана». Эур Дж Фармакол . 482 (1–3): 9–16. дои : 10.1016/j.ejphar.2003.09.044 . ПМИД 14659999 .
- ^ Jump up to: а б с Харсинг Л.Г., Нолл Дж., Микля И. (август 2022 г.). «Усилитель регуляции дофаминергической нейрохимической передачи в полосатом теле» . Int J Mol Sci . 23 (15): 8543. doi : 10.3390/ijms23158543 . ПМЦ 9369307 . ПМИД 35955676 .
- ^ Jump up to: а б Пей Ю, Асиф-Малик А, Каналес Дж.Дж. (2016). «Следовые амины и рецептор 1, связанный с следовыми аминами: фармакология, нейрохимия и клинические последствия» . Передние нейроны . 10 : 148. дои : 10.3389/fnins.2016.00148 . ПМЦ 4820462 . ПМИД 27092049 .
- ^ Jump up to: а б Рутильяно Дж., Аккоррони А., Зукки Р. (2017). «Дело в пользу TAAR1 как модулятора функции центральной нервной системы» . Фронт Фармакол . 8 : 987. дои : 10.3389/fphar.2017.00987 . ПМК 5767590 . ПМИД 29375386 .
- ^ Мадьяр К., Сзенде Б., Дженей В., Таби Т., Палфи М., Сёко Э. (декабрь 2010 г.). «R-депренил: фармакологический спектр активности». Нейрохим Рез . 35 (12): 1922–1932. дои : 10.1007/s11064-010-0238-8 . ПМИД 20725780 .
- ^ Jump up to: а б Нолл Дж., Йонеда Ф., Нолл Б., Оде Х., Микля I (декабрь 1999 г.). «(-)1-(Бензофуран-2-ил)-2-пропиламинопентан, [(-)BPAP], селективный усилитель опосредованного распространением импульса высвобождения катехоламинов и серотонина в мозге» . Бр Джей Фармакол . 128 (8): 1723–1732. дои : 10.1038/sj.bjp.0702995 . ПМК 1571822 . ПМИД 10588928 .
- ^ Экблом Дж., Ореланд Л., Чен К., Ши Дж.К. (1998). «Существует ли макромолекулярная мишень для L-депренила, не являющаяся МАО?: Исследования на мутантных мышах MAOB». Наука о жизни . 63 (12): PL181–6. дои : 10.1016/s0024-3205(98)00370-1 . ПМИД 9749831 .
- ^ Сотникова Т.Д., Карон М.Г., Гайнетдинов Р.Р. (август 2009 г.). «Отследить аминоассоциированные рецепторы как новые терапевтические мишени» . Мол Фармакол . 76 (2): 229–235. дои : 10.1124/моль.109.055970 . ПМЦ 2713119 . ПМИД 19389919 .
- ^ Риз Э.А., Норимацу Ю., Гранди М.С., Сучленд К.Л., Бунцов-младший, Гранди Д.К. (январь 2014 г.). «Изучение факторов, определяющих функциональную селективность рецептора 1, связанного со следами аминов, в отношении стереоизомеров амфетамина и метамфетамина». J Med Chem . 57 (2): 378–390. дои : 10.1021/jm401316v . ПМИД 24354319 .
- ^ «Левметамфетамин» . ПабХим . Национальный центр биотехнологической информации, Национальная медицинская библиотека США. Архивировано из оригинала 18 октября 2014 года . Проверено 17 октября 2014 г.
- ^ Jump up to: а б Нолл Дж., Микля И., Нолл Б., Марко Р., Келемен К. (1996). «(-) Депренил и (-) 1-фенил-2-пропиламинопентан, [(-) PPAP], действуют прежде всего как мощные стимуляторы взаимодействия потенциала действия и высвобождения медиатора в катехоламинергических нейронах». Наука о жизни . 58 (10): 817–827. дои : 10.1016/0024-3205(96)00014-8 . ПМИД 8602114 .
- ^ Jump up to: а б Нолл Дж., Микля I (1994). «Многократное введение небольших доз (-) депренила усиливает катехоламинергическую активность и снижает серотонинергическую активность в головном мозге, и эти эффекты не связаны с ингибированием МАО-В». Арх Инт Фармакодин Тер . 328 (1): 1–15. ПМИД 7893186 .
- ^ Jump up to: а б с д и Микля I (июнь 2014 г.). «Существенная разница между фармакологическим спектром (-)-депренила и разагилина». Представитель Фармакол . 66 (3): 453–458. дои : 10.1016/j.pharep.2013.11.003 . ПМИД 24905523 .
- ^ Микля I (март 2008 г.). Сравнение фармакологии (-)-депренила, N-метилпрогариламин-1-аминоиндана (J-508) и десметилового аналога J-508 (разагилина)» [Сравнение фармакологии (-)-депренила с N -метилпропаргиламин-1-аминоиндан (J-508) и разагилин, десметил-аналог J-508] (PDF) . Нейропсихофармакол Хунг (на венгерском языке). 10 (1): 15–22. ПМИД 18771016 .
- ^ Jump up to: а б Брэди Л.С., Лисанби С.Х., Гордон Дж.А. (2023). «Новые направления в разработке психиатрических препаратов: многообещающие методы лечения в стадии разработки». Экспертное мнение о лекарствах . 18 (8): 835–850. дои : 10.1080/17460441.2023.2224555 . ПМИД 37352473 .
- ^ Jump up to: а б Куварзин С.Р., Суханов И., Онохин К., Захаров К., Гайнетдинов Р.Р. (июль 2023 г.). «Раскрытие терапевтического потенциала улотаронта как агониста аминоассоциированного рецептора 1 при нервно-психических расстройствах» . Биомедицины . 11 (7): 1977. doi : 10.3390/biomedicines11071977 . ПМЦ 10377193 . ПМИД 37509616 .
- ^ Jump up to: а б Далло Дж., Лекка Н., Нолл Дж. (1986). «Эякуляторное поведение сексуально вялых самцов крыс, получавших (-) депренил, апоморфин, бромокриптин и амфетамин». Пол Джей Фармакол Фарм . 38 (3): 251–255. ПМИД 3095802 .
- ^ Jump up to: а б Йен Т.Т., Далло Дж., Нолл Дж. (1982). «Афродизиакальный эффект низких доз (-) депренила у крыс-самцов». Пол Джей Фармакол Фарм . 34 (5–6): 303–308. ПМИД 6821215 .
- ^ Jump up to: а б Нолл Дж., Далло Дж., Йен Т.Т. (1989). «Стриатальный дофамин, сексуальная активность и продолжительность жизни. Продолжительность жизни крыс, получавших (-) депренил». Наука о жизни . 45 (6): 525–531. дои : 10.1016/0024-3205(89)90103-3 . ПМИД 2505007 .
- ^ Чемберс К.К., Феникс CH (август 1989 г.). «Апоморфин, депренил и йохимбин не способны повысить сексуальное поведение у мужчин-резусов». Поведение нейробиологов . 103 (4): 816–823. дои : 10.1037/0735-7044.103.4.816 . ПМИД 2504225 .
- ^ Jump up to: а б с д и ж Heal DJ, Smith SL, Gosden J, Nutt DJ (июнь 2013 г.). «Амфетамин, прошлое и настоящее – фармакологическая и клиническая перспектива» . Дж Психофармакол . 27 (6): 479–496. дои : 10.1177/0269881113482532 . ПМК 3666194 . ПМИД 23539642 .
- ^ Jump up to: а б с Фернандес Х.Х., Чен Дж.Дж. (декабрь 2007 г.). «Ингибирование моноаминоксидазы-В при лечении болезни Паркинсона». Фармакотерапия . 27 (12 ПТ 2): 174С–185С. дои : 10.1592/phco.27.12part2.174S . ПМИД 18041937 .
- ^ Мюллер Т., Хоффманн Дж.А., Димпфель В., Ольвайн С. (май 2013 г.). «Переход с селегилина на разагилин полезен у пациентов с болезнью Паркинсона». J Neural Transm (Вена) . 120 (5): 761–765. дои : 10.1007/s00702-012-0927-3 . ПМИД 23196982 .
- ^ Ринальди Д., Альборгетти М., Бьянкини Е., Сфорца М., Галли С., Понтьери Ф.Е. (2023). «Ингибиторы моноаминоксидазы типа B и когнитивные функции при болезни Паркинсона: помимо основного механизма действия» . Карр Нейрофармакол . 21 (5): 1214–1223. дои : 10.2174/1570159X20666220905102144 . ПМЦ 10286595 . ПМИД 36065929 .
- ^ Энгберг Г., Элебринг Т., Ниссбрандт Х. (ноябрь 1991 г.). «Депренил (селегилин), селективный ингибитор МАО-В с активными метаболитами; влияние на двигательную активность, дофаминергическую нейротрансмиссию и скорость срабатывания нигральных дофаминовых нейронов». Журнал фармакологии и экспериментальной терапии . 259 (2): 841–847. ПМИД 1658311 .
- ^ Бундгаард С, Монтезиньо ЛП, Андерсон Н, Томсен С, Мёрк А (2016). «Селегилин вызывает эффект пробуждения у крыс, связанный с образованием его активных метаболитов». Фармакол Биохим Поведение . 150–151: 147–152. дои : 10.1016/j.pbb.2016.10.003 . ПМИД 27984094 .
- ^ Jump up to: а б Симпсон Л.Л. (1978). «Доказательства того, что депренил, ингибитор моноаминоксидазы типа B, является симпатомиметическим амином косвенного действия». Биохим Фармакол . 27 (11): 1591–1595. дои : 10.1016/0006-2952(78)90490-2 . ПМИД 697901 .
- ^ Ротман Р.Б., Бауманн М.Х., Дерш К.М., Ромеро Д.В., Райс К.С., Кэрролл Ф.И. и др. (январь 2001 г.). «Стимуляторы центральной нервной системы амфетаминового типа высвобождают норадреналин более эффективно, чем дофамин и серотонин». Синапс . 39 (1): 32–41. doi : 10.1002/1098-2396(20010101)39:1<32::AID-SYN5>3.0.CO;2-3 . ПМИД 11071707 .
- ^ Jump up to: а б с Кохут С.Дж., Джейкобс Д.С., Ротман Р.Б., Партилла Дж.С., Бергман Дж., Блау Б.Е. (декабрь 2017 г.). «Кокаиноподобные дискриминационные стимулирующие эффекты «предпочитающих норэпинефрин» моноаминовых релизеров: временной ход и исследования взаимодействия на макаках-резусах» . Психофармакология (Берл) . 234 (23–24): 3455–3465. дои : 10.1007/s00213-017-4731-5 . ПМЦ 5747253 . ПМИД 28889212 .
- ^ Jump up to: а б с д Мендельсон Дж., Уэмура Н., Харрис Д., Нат Р.П., Фернандес Э., Джейкоб П. и др. (октябрь 2006 г.). «Человеческая фармакология стереоизомеров метамфетамина». Клин Фармакол Тер . 80 (4): 403–420. дои : 10.1016/j.clpt.2006.06.013 . ПМИД 17015058 .
- ^ Jump up to: а б Сюэ З, Семиан Дж. Н., Чжу Ц, Блаф Б. Е., Ли Дж. Х. (август 2019 г.). «Дальнейшее фармакологическое сравнение D-метамфетамина и L-метамфетамина у крыс: поведенческие и физиологические показатели, связанные со злоупотреблением» . Бехав Фармакол . 30 (5): 422–428. дои : 10.1097/FBP.0000000000000453 . ПМК 6529304 . ПМИД 30480551 .
- ^ Jump up to: а б с д и ж г час Нишино С, Котории Н (2016). «Способы действия препаратов, связанных с нарколепсией: фармакология соединений, способствующих пробуждению, и антикатаплектических средств». Нарколепсия: Клиническое руководство (2-е изд.). Чам: Международное издательство Springer. стр. 307–329. дои : 10.1007/978-3-319-23739-8_22 . ISBN 978-3-319-23738-1 .
- ^ Jump up to: а б с Кученски Р., Сигал Д.С., Чо А.К., Мелега В. (февраль 1995 г.). «Норадреналин в гиппокампе, хвостатый дофамин и серотонин, а также поведенческие реакции на стереоизомеры амфетамина и метамфетамина» . Дж. Нейроски . 15 (2): 1308–1317. doi : 10.1523/JNEUROSCI.15-02-01308.1995 . ПМК 6577819 . ПМИД 7869099 .
- ^ Нисимура Т., Такахата К., Косуги Ю., Танабэ Т., Мураока С. (май 2017 г.). «Различия в психомоторном эффекте между l-метамфетамином и d-метамфетамином не зависят от фармакокинетических профилей мышиной плазмы и мозга» . J Neural Transm (Вена) . 124 (5): 519–523. дои : 10.1007/s00702-017-1694-y . ПМК 5399046 . ПМИД 28213761 .
- ^ Симиан Дж. Н., Сюэ З., Блаф Б. Е., Ли Дж. Х. (июль 2017 г.). «Сравнение некоторых поведенческих эффектов d- и l-метамфетамина у взрослых крыс-самцов» . Психофармакология (Берл) . 234 (14): 2167–2176. дои : 10.1007/s00213-017-4623-8 . ПМЦ 5482751 . ПМИД 28386698 .
- ^ Поли Р.К., Бхимани Р.В., Ли Дж.К., Блаф Б.Е., Ландавазо А., Пак Дж. (март 2023 г.). «Отличительные эффекты изомеров метамфетамина на лимбическую передачу норадреналина и дофамина в мозге крысы». ACS Химическая Нейронаука : acschemneuro.2c00689. дои : 10.1021/acschemneuro.2c00689 . ПМИД 36976755 . S2CID 257772503 .
- ^ Jump up to: а б с д и Смит Р.К., Дэвис Дж.М. (июнь 1977 г.). «Сравнительное влияние d-амфетамина, l-амфетамина и метилфенидата на настроение человека». Психофармакология (Берл) . 53 (1): 1–12. дои : 10.1007/BF00426687 . ПМИД 407607 .
- ^ Jump up to: а б с д Бархольц Х.М., Хадзима Р., Майлз А. (июль 2023 г.). «Фармакология R-(-)-метамфетамина у людей: систематический обзор литературы» . ACS Pharmacol Transl Sci . 6 (7): 914–924. дои : 10.1021/acptsci.3c00019 . ПМЦ 10353062 . ПМИД 37470013 .
- ^ Jump up to: а б Ли Л, Лопес Дж. К., Галлоуэй Г.П., Бэгготт М.Дж., Эверхарт Т., Мендельсон Дж. (август 2010 г.). «Оценка потребления злоупотребляемых метамфетаминов с использованием введенного экспериментатором дейтерия, меченного R-метамфетамином: выбор дозы R-метамфетамина» . The Drug Monit . 32 (4): 504–507. дои : 10.1097/FTD.0b013e3181db82f2 . ПМК 3040572 . ПМИД 20592647 .
- ^ Jump up to: а б Сильверстоун Т., Уэллс Б. (1980). «Клиническая психофармакология амфетамина и родственных соединений». Амфетамины и родственные стимуляторы: химические, биологические, клинические и социологические аспекты . ЦРК Пресс. стр. 147–160. дои : 10.1201/9780429279843-10 . ISBN 978-0-429-27984-3 .
- ^ Паркс Дж. Д., Фентон Г. В. (декабрь 1973 г.). «Лево(-) амфетамин и декстро(+) амфетамин в лечении нарколепсии» . J Neurol Нейрохирургия Психиатрия . 36 (6): 1076–1081. дои : 10.1136/jnnp.36.6.1076 . ПМЦ 1083612 . ПМИД 4359162 .
- ^ Jump up to: а б с Паркс Дж.Д., Тарси Д., Марсден К.Д., Бовилл К.Т., Фиппс Дж.А., Роуз П. и др. (март 1975 г.). «Амфетамины в лечении болезни Паркинсона» . J Neurol Нейрохирургия Психиатрия . 38 (3): 232–237. дои : 10.1136/jnnp.38.3.232 . ПМК 491901 . ПМИД 1097600 .
- ^ Jump up to: а б Элсворт Дж. Д., Сэндлер М., Лиз А. Дж., Уорд С., Стерн Г. М. (1982). «Вклад метаболитов амфетамина (-)-депренила в его противопаркинсонические свойства». J Нейронная передача . 54 (1–2): 105–110. дои : 10.1007/BF01249283 . ПМИД 6809891 .
- ^ Jump up to: а б Маленка Р.Ц., Нестлер Э.Дж., Хайман С.Е., Хольцман Д.М. (2015). «Глава 16: Подкрепление и аддиктивные расстройства». Молекулярная нейрофармакология: фонд клинической неврологии (3-е изд.). Нью-Йорк: McGraw-Hill Medical. ISBN 9780071827706 .
В отличие от кокаина и амфетамина, метамфетамин непосредственно токсичен в более высоких дозах для дофаминовых нейронов среднего мозга.
- ^ Jump up to: а б Мощинска А., Каллан С.П. (сентябрь 2017 г.). «Молекулярные, поведенческие и физиологические последствия нейротоксичности метамфетамина: последствия для лечения» . Журнал фармакологии и экспериментальной терапии . 362 (3): 474–488. дои : 10.1124/jpet.116.238501 . ПМК 11047030 . ПМИД 28630283 .
В отчете Национальной администрации безопасности дорожного движения США говорится, что «чистота МЕТА в настоящее время очень высока, 60–90%», т. е. незаконный МЕТ представляет собой преимущественно d-МЕТН, и что «обычные дозы, которыми злоупотребляют, составляют 100–1000 мг». /день и до 5000 мг/день при хроническом злоупотреблении». Результаты нескольких опросов и исследований хронического злоупотребления метамфетамином в Соединенных Штатах согласуются с этим отчетом; в среднем, по данным самооценки, хроническое употребление метамфетамина составляло 0,25–1,6 г/день. ...
Кроме того, хронические потребители метана могут подвергаться более высокому риску развития болезни Паркинсона (БП), чем те, кто не употребляет его, из-за токсического воздействия препарата на нигростриарный путь DA. ...
Имеются доказательства того, что L-изомер метана или амфетамина в большей степени способствует периферическим эффектам (например, частоте сердечных сокращений), а d-изомер больше способствует эффектам на ЦНС (например, эйфории). - ^ Чжоу З.Д., И Л.С., Ван Д.К., Лим Т.М., Тан ЭК (сентябрь 2023 г.). «Роль дофамина в патофизиологии болезни Паркинсона» . Трансляционная нейродегенерация . 12 (1): 44. дои : 10.1186/s40035-023-00378-6 . ПМЦ 10506345 . ПМИД 37718439 .
Прогрессирующая дегенерация дофаминергических нейронов снижает содержание ДА в ЧС и полосатом теле и провоцирует возникновение клинических симптомов БП, таких как тремор, постуральная нестабильность, брадикинезия и мышечная ригидность.
- ^ Jump up to: а б Дези Л., Весей Л. (2017). «Ингибиторы моноаминоксидазы B при болезни Паркинсона». Цели применения лекарств при расстройствах нейронов ЦНС . 16 (4): 425–439. дои : 10.2174/1871527316666170124165222 . ПМИД 28124620 .
- ^ Jump up to: а б с д и ж Нолл Дж (1995). «Обоснование применения (-) депренила (селегилина) при болезни Паркинсона и профилактике возрастных изменений нигрела». Биомед Фармакотер . 49 (4): 187–195. дои : 10.1016/0753-3322(96)82619-9 . ПМИД 7669938 .
- ^ Jump up to: а б с д и ж Нолл Дж (1993). «Фармакологическая основа терапевтического эффекта (—)-депренила при возрастных неврологических заболеваниях». Ингибиторы моноаминоксидазы B: фармакология и клиническое применение при нейродегенеративных заболеваниях . Вехи в лекарственной терапии. Базель: Биркхойзер Базель. стр. 145–168. дои : 10.1007/978-3-0348-6348-3_7 . ISBN 978-3-0348-6349-0 .
- ^ Jump up to: а б с д и Герлах М., Ридерер П. (1993). «Патофизиологические основы болезни Паркинсона». Ингибиторы моноаминоксидазы B: фармакология и клиническое применение при нейродегенеративных заболеваниях . Вехи в лекарственной терапии (на немецком языке). Базель: Биркхойзер Базель. стр. 25–50. дои : 10.1007/978-3-0348-6348-3_2 . ISBN 978-3-0348-6349-0 .
- ^ Ридерер П., Вукетич С. (1976). «Временной ход нигростриарной дегенерации при болезни Паркинсона. Детальное исследование влиятельных факторов в анализе аминов в мозге человека». J Нейронная передача . 38 (3–4): 277–301. дои : 10.1007/BF01249445 . ПМИД 956814 .
- ^ Нолл Дж. (август 1994 г.). «Воспоминания о моих 45 годах исследований». Фармакол Токсикол . 75 (2): 65–72. дои : 10.1111/j.1600-0773.1994.tb00326.x . ПМИД 7971740 .
- ^ Jump up to: а б с д и Хили Д. (2000). «Психофармакология жизни и смерти. Интервью с Джозефом Ноллом». Психофармакологи, Vol. III: Интервью . Лондон: Арнольд. стр. 81–110. ISBN 978-0-340-76110-6 .
- ^ МоханКумар П.С., МоханКумар С.М., Квадри СК (апрель 2001 г.). «Депренил стимулирует отток моноаминов из гипоталамуса крысы in vitro». Мозговой Рес Булл . 54 (6): 675–680. дои : 10.1016/s0361-9230(01)00481-6 . ПМИД 11403995 .
- ^ Ицхак Ю. (1994). Сигма-рецепторы . Академическая пресса. стр. 84–. ISBN 978-0-12-376350-1 .
- ^ Стоун Т.В. (1993). Ацетилхолин, сигма-рецепторы, CCK и эйкозаноиды, нейротоксины . Тейлор и Фрэнсис. стр. 124–. ISBN 978-0-7484-0063-8 .
- ^ Jump up to: а б Дефтереос С.Н., Андронис Калифорния (июль 2010 г.). «Диссогласующиеся эффекты доз разагилина при болезни Паркинсона». Нат преподобный Нейрол . 6 (7): 1п после 410. doi : 10.1038/nrneurol.2010.2-c1 . ПМИД 20653097 .
- ^ Jump up to: а б Таттон В., Чалмерс-Редман Р., Таттон Н. (май 2003 г.). «Нейропротекция депренилом и другими пропаргиламинами: глицеральдегид-3-фосфатдегидрогеназа, а не моноаминоксидаза B». J Neural Transm (Вена) . 110 (5): 509–515. дои : 10.1007/s00702-002-0827-z . ПМИД 12721812 .
- ^ Аззаро А.Дж., Зиемняк Дж., Кемпер Э., Кэмпбелл Б.Дж., ВанДенберг С. (октябрь 2007 г.). «Фармакокинетика и абсолютная биодоступность селегилина после лечения здоровых людей трансдермальной системой селегилина (6 мг/24 часа): сравнение с пероральными капсулами селегилина». Джей Клин Фармакол . 47 (10): 1256–1267. дои : 10.1177/0091270007304779 . ПМИД 17715422 .
- ^ Пас-Рамос М.И., Круз С.Л., Виоланте-Сориа В (2023). «Стимуляторы амфетаминового ряда: новый взгляд на их действие и закономерности использования» . Рев Инвест Клин . 75 (3): 143–157. дои : 10.24875/RIC.23000110 . ПМИД 37441770 .
- ^ Барретт Дж.С., Сего П., Рохатаги С., Моралес Р.Дж., Де Витт К.Э., Раевски Г. и др. (октябрь 1996 г.). «Абсорбция и пресистемный метаболизм селегилина гидрохлорида в различных отделах желудочно-кишечного тракта у здоровых мужчин». Фармацевтические исследования . 13 (10): 1535–1540. дои : 10.1023/А:1016035730754 . ПМИД 8899847 . S2CID 24654277 .
- ^ Мусшофф Ф. (февраль 2000 г.). «Незаконное или законное использование? Соединения-прекурсоры амфетамина и метамфетамина». Препарат Метаб Ред . 32 (1): 15–44. дои : 10.1081/dmr-100100562 . ПМИД 10711406 .
- ^ Коди Джей Ти (май 2002 г.). «Прекурсоры как источник положительных результатов тестирования на наркотики метамфетамина и/или амфетамина». J Occup Environ Med . 44 (5): 435–450. дои : 10.1097/00043764-200205000-00012 . ПМИД 12024689 .
- ^ Веленоси Т.Дж., Уркхарт Б.Л. (август 2014 г.). «Фармакокинетические соображения при хронической болезни почек и у пациентов, нуждающихся в диализе». Экспертное мнение о препарате Метаб Токсикол . 10 (8): 1131–1143. дои : 10.1517/17425255.2014.931371 . ПМИД 24961255 .
- ^ Jump up to: а б Багдади Н.Т., Баник С., Шварц С.А., Макинтайр Р.С. (апрель 2009 г.). «Психотропные препараты и почечная недостаточность: применение данных для клинической практики». Адв Тер . 26 (4): 404–424. дои : 10.1007/s12325-009-0021-x . ПМИД 19444657 .
- ^ Милиус В., Мёллер Х.К., Болхальтер С., Чампи де Андраде Д., Перес Льорет С. (июль 2021 г.). «Диагностика и лечение боли при болезни Паркинсона: новый подход». Наркотическое старение . 38 (7): 559–577. дои : 10.1007/s40266-021-00867-1 . ПМИД 34224103 .
- ^ Jump up to: а б с «Селегилин» . ПабХим . Проверено 18 июля 2024 г.
- ^ Jump up to: а б с «Селегилин: использование, взаимодействие, механизм действия» . ДругБанк Онлайн . 5 июня 1989 года . Проверено 18 июля 2024 г.
- ^ Jump up to: а б с «Селегилин» . Химический Паук . 21 июля 2022 г. Проверено 18 июля 2024 г.
- ^ Jump up to: а б с д и Швейцарская ассоциация фармацевтов (2000). Index Nominum 2000: Международный каталог лекарств . Научные издательства Медфарм. п. 939. ИСБН 978-3-88763-075-1 . Проверено 4 июля 2024 г.
- ^ Фаулер С.Дж., Ореланд Л., Каллингем, Б.А. (июнь 1981 г.). «Ингибиторы ацетиленовой моноаминоксидазы хлоргилин, депренил, паргилин и J-508: их свойства и применение». Джей Фарм Фармакол . 33 (6): 341–347. дои : 10.1111/j.2042-7158.1981.tb13800.x . ПМИД 6115003 .
- ^ Ульрих С., Рикен Р., Адли М. (август 2017 г.). «Транилципромин в виду (Часть I): Обзор фармакологии» . Eur Нейропсихофармакол . 27 (8): 697–713. дои : 10.1016/j.euroneuro.2017.05.007 . ПМИД 28655495 .
- ^ Рикен Р., Ульрих С., Шлаттманн П., Адли М. (август 2017 г.). «Транилципромин в виду (Часть II): обзор клинической фармакологии и метаанализ контролируемых исследований депрессии» . Eur Нейропсихофармакол . 27 (8): 714–731. дои : 10.1016/j.euroneuro.2017.04.003 . ПМИД 28579071 .
- ^ Jump up to: а б Мадьяр К (1994). «Поведение (-)-депренила и его аналогов». J Приложение для нейронной передачи . 41 : 167–175. дои : 10.1007/978-3-7091-9324-2_23 . ISBN 978-3-211-82521-1 . ПМИД 7931223 .
- ^ Нолл Дж., Эчери З., Мадьяр К., Сатори Э. (1978). «Новые (-) производные депренила селективные ингибиторы моноаминоксидазы B-типа. Связь структуры с их действием». Биохим Фармакол . 27 (13): 1739–1747. дои : 10.1016/0006-2952(78)90550-6 . ПМИД 708454 .
- ^ Мадьяр К., Эчери З., Бернат Г., Сатори Э., Нолл Дж. (1980). «Взаимосвязь структура-активность селективных ингибиторов МАО-Б». Моноаминоксидазы и их избирательное ингибирование . Пергамон. стр. 11–21.
- ^ Нолл Дж (1980). «Селективные ингибиторы МАО-Б с различным фармакологическим профилем». Моноаминоксидазы и их избирательное ингибирование . Пергамон. стр. 23–36.
- ^ Вайнреб О., Амит Т., Бар-Ам О., Юдим М.Б. (апрель 2012 г.). «Ладостигил: новый мультимодальный нейропротекторный препарат с активностью ингибирования холинэстеразы и селективной моноаминоксидазы в мозге для лечения болезни Альцгеймера». Цели Curr по борьбе с наркотиками . 13 (4): 483–494. дои : 10.2174/138945012799499794 . ПМИД 22280345 .
- ^ DE 1568277 , Ecsery Z, Kosa I, Knoll J, Somfai E, "Verfahren zur Herstellung von neuen,optisch aktiven Phenylisopylamin-Derivaten [Способ получения новых оптически активных производных фенилизопиламина]", опубликовано 30 апреля 1970 г., присвоено на завод фармацевтической и химической продукции «Чиноин» РТ
- ^ Дж. Герман Ни Воероэс, З. Эксери, Г. Сабо, Л. Арваи, Л. Надь, О. Орбан, Э. Санфаи, патент США 4 564 706 (1986).
- ^ EP 344675 , Hájicek J, Hrbata J, Pihera P, Brunova B, Ferenc M, Krepelka J, Kvapil L, Pospisil J, «Способ производства гидрохлорида селегилина», опубликовано 989-12-06, передано SPOFA Spojené Podniky Pro Медицинская продукция
- ^ Фаулер Дж. С. (июль 1977 г.). «2-Метил-3-бутин-2-ол как предшественник ацетилена в реакции Манниха. Новый синтез суицидных инактиваторов моноаминоксидазы». Журнал органической химии . 42 (15): 2637–2639. дои : 10.1021/jo00435a026 . ПМИД 874623 .
- ^ Целлер Э.А., Барский Дж (ноябрь 1952 г.). «Ингибирование моноаминоксидазы печени и мозга in vivo 1-изоникотинил-2-изопропилгидразином». Proc Soc Exp Biol Med . 81 (2): 459–461. дои : 10.3181/00379727-81-19910 . ПМИД 13027339 .
- ^ «Санофи расширяет холдинг в Чиноине» . Фармацевтическое письмо . 19 сентября 1993 года.
- ^ Jump up to: а б с д и Нолл Дж., Эчери З., Келемен К., Нивель Дж., Нолл Б. (май 1965 г.). «Фенилизопропилметилпропиниламин (Е-250), психический энергетик нового спектра». Archives Internationales de Pharmacodynamie et de Therapie . 155 (1): 154–164. ПМИД 4378644 .
- ^ Брайант Дж.М., Торосдаг С., Шварц Н., Флетчер Л., Фертиг Х., Шварц М.С. и др. (октябрь 1961 г.). «Антигипертензивные свойства паргилина гидрохлорида. Новый негидразиновый ингибитор моноаминоксидазы по сравнению с сульфонамидными диуретиками». ДЖАМА . 178 : 406–409. дои : 10.1001/jama.1961.73040430005010 . ПМИД 13874134 .
- ^ Джонстон Дж. П. (июль 1968 г.). «Некоторые наблюдения о новом ингибиторе моноаминоксидазы в тканях головного мозга». Биохим Фармакол . 17 (7): 1285–1297. дои : 10.1016/0006-2952(68)90066-x . ПМИД 5659776 .
- ^ Нолл Дж., Мадьяр К. (1972). «Некоторые загадочные фармакологические эффекты ингибиторов моноаминоксидазы». Адв Биохим Психофармакол . 5 : 393–408. ПМИД 5066229 .
- ^ Нолл Дж., Визи Э.С., Сомоджи Г. (1968). «Фенилизопропилметилпропиниламин (Е-250), ингибитор моноаминоксидазы, противодействующий действию тирамина» . Исследования наркотиков . 18 (1): 109–112.
- ^ Варга Э (1965). «Предварительный отчет о действии препарата Е-250 (фенил-изопропил-метил-пропиниламина хлоргидрат)». III Hungarica conferentia pro Therapia et Investigatione in Pharmacologia . Будапешт: Издательство Венгерской академии наук. стр. 197–201.
- ^ Jump up to: а б Варга Э., Трингер Л. (1967). «Клиническое исследование нового типа психоэнергетического средства быстрого действия (фенил-изопропил-метилпропинил-HCl, «Е-250»)». Acta Med Acad Sci Hung . 23 (3): 289–295. ПМИД 6056555 .
- ^ Трингер Л., Хайтс Г., Варга Э. (1971). «Эффект (-) E-250, (-) L-фенилизопропилметилпропиниламина HCl при депрессии». 5-я Венгерская конференция по терапии и исследованиям в области фармакологии . стр. 111–114.
- ^ Манн Дж., Гершон С. (март 1980 г.). «L-депренил, селективный ингибитор моноаминоксидазы типа B при эндогенной депрессии». Наука о жизни . 26 (11): 877–882. дои : 10.1016/0024-3205(80)90350-1 . ПМИД 6768943 .
- ^ Jump up to: а б Биркмайер В., Ридерер П., Линауэр В., Нолл Дж. (1984). «L-депренил плюс L-фенилаланин в лечении депрессии». J Нейронная передача . 59 (1): 81–87. дои : 10.1007/BF01249880 . ПМИД 6425455 .
- ^ Биркмайер В., Ридерер П., Юдим М.Б., Линауэр В. (1975). «Потенцирование антиакинетического эффекта после лечения леводопой ингибитором МАО-В Депренилом» . Журнал нейронной передачи . 36 (3–4): 303–326. дои : 10.1007/BF01253131 . ПМИД 1172524 . S2CID 38179089 . Архивировано из оригинала 12 февраля 2013 года.
- ^ Jump up to: а б Кроми WJ (7 ноября 2002 г.). «Бодкин лечит депрессию» . Вестник Гарвардского университета . Проверено 8 сентября 2007 г.
- ^ Рейнольдс Г.П., Элсворт Дж.Д., Блау К., Сэндлер М., Лиз А.Дж., Стерн ГМ (декабрь 1978 г.). «Депренил у человека метаболизируется до метамфетамина и амфетамина» . Бр Дж Клин Фармакол . 6 (6): 542–544. дои : 10.1111/j.1365-2125.1978.tb00883.x . ПМЦ 1429688 . ПМИД 728327 .
- ^ Нолл Дж., Нолл Б., Тёрёк З., Тимар Дж., Ясар С. (1992). «Фармакология 1-фенил-2-пропиламинопентана (PPAP), психостимулятора нового спектра действия, полученного из депренила». Арх Инт Фармакодин Тер . 316 : 5–29. ПМИД 1356324 .
- ^ Гаснер П., Микля I (декабрь 2004 г.). «Использование синтетических усилителей (-)-депренила и (-)-BPAP при большой депрессии». Нейропсихофармакол Хунг . 6 (4): 210–220. ПМИД 15825677 .
- ^ Фрэмптон Дж. Э., Плоскер Г. Л. (2007). «Трансдермальная система Селегилин: в лечении большого депрессивного расстройства». Наркотики . 67 (2): 257–265, обсуждение 266–267. дои : 10.2165/00003495-200767020-00006 . ПМИД 17284087 . S2CID 42425086 .
- ^ Даффи М. (3 декабря 2002 г.). «Патч вселяет новую надежду на победу над депрессией» . Нью-Йорк Таймс . ISSN 0362-4331 .
- ^ Каскад Э.Ф., Калали А.Х., Прескорн С.Х. (июнь 2007 г.). «Эмсам: первый год» . Психиатрия . 4 (6): 19–21. ПМЦ 2921248 . ПМИД 20711332 .
- ^ Jump up to: а б с Элкс Дж. (1990). Словарь лекарств: Химические данные: Химические данные, структуры и библиография . Спрингер США. п. 441. ИСБН 978-1-4757-2085-3 . Проверено 4 июля 2024 г.
- ^ Jump up to: а б Мортон И.К., Холл Дж.М. (1999). Краткий словарь фармакологических средств: свойства и синонимы . Спрингер Нидерланды. п. 254. ИСБН 978-94-011-4439-1 . Проверено 4 июля 2024 г.
- ^ Фейнберг С. (1 ноября 2006 г.). «EMSAM: удобный MAOI?» . КАРЛАТ ПУБЛИШИНГ . Проверено 4 июля 2024 г.
- ^ Дорси Э.Р., Томпсон Дж.П., Даюб Э.Дж., Джордж Б., Зауберманн Л.А., Холлоуэй Р.Г. (июль 2009 г.). «Дефицит селегилина: причины и издержки нехватки непатентованных лекарств» . Неврология . 73 (3): 213–217. дои : 10.1212/WNL.0b013e3181ae7b04 . ПМЦ 2715573 . ПМИД 19620609 .
- ^ Jump up to: а б Фердинанди П., Йонеда Ф., Мураока С., Фюрст С., Гиреш К., Микля И. (февраль 2020 г.). «Геропротекция в будущем. Памяти Джозефа Нолла: история селегилина продолжается» . Европейский журнал фармакологии . 868 : 172793. doi : 10.1016/j.ejphar.2019.172793 . ПМИД 31743738 . S2CID 208185366 .
- ^ Пирс Д. (1995). Гедонистический императив . OCLC 44325836 .
- ^ «Сэм Баркер и Дэвид Пирс об искусстве, райской инженерии и экзистенциальной надежде (с гостевым миксом) | Подкаст FLI» . Институт будущего жизни (аудио, стенограмма). 24 июня 2020 г.
- ^ Jump up to: а б Мерфи HT (14 декабря 2022 г.). «Сэм Бэнкман-Фрид подтвердил, что носит нашивку Emsam. Что такое нашивка Emsam?» . Журнал «Сланец» . Проверено 2 июля 2024 г.
- ^ Jump up to: а б Сигалос М. (14 августа 2023 г.). «Сэм Бэнкман-Фрид получает разрешение на прием Аддералла от СДВГ, находясь в тюрьме» . CNBC . Проверено 2 июля 2024 г.
- ^ Александр С (16 ноября 2022 г.). «Психофармакология краха FTX» . Астральный Кодекс Десять . Проверено 4 июля 2024 г.
- ^ Гурвиц Г. (2019). Из темноты . Книги о пингвинах. п. 431. ИСБН 9780718185480 .
- ^ Лидский Т., Шнайдер Дж. (2010). Конфеты для мозга: увеличьте мощность своего мозга с помощью витаминов, пищевых добавок, лекарств и других веществ . Пробный камень. стр. 89–93. ISBN 978-0-7432-1843-6 . Проверено 5 июля 2024 г.
- ^ Jump up to: а б Микля I (2011). «Концепция холмика по снижению распространенности болезни Паркинсона». В Финкельштейне Д. (ред.). На пути к новым методам лечения болезни Паркинсона . стр. 77–100. ISBN 978-953-307-463-4 . Проверено 29 июля 2024 г.
- ^ Сондерс Н., Херон Л. (1993). Е значит экстази . Лондон: Н. Сондерс. ISBN 978-0-9501628-8-1 . OCLC 29388575 . [ нужна страница ]
- ^ Сондерс Н. «Результаты испытаний 30 образцов экстази, купленных в британских клубах в период с 11.94 по 7.95» .
- ^ Jump up to: а б с д Дочерти-младший (июнь 2008 г.). «Фармакология стимуляторов, запрещенных Всемирным антидопинговым агентством (ВАДА)» . Бр Джей Фармакол . 154 (3): 606–622. дои : 10.1038/bjp.2008.124 . ПМЦ 2439527 . ПМИД 18500382 .
- ^ Чжоу С.Ф., Лю Дж.П., Лай XS (2009). «Субстратная специфичность, ингибиторы и регуляция человеческого цитохрома P450 2D6 и значение для разработки лекарств». Curr Med Chem . 16 (21): 2661–805. дои : 10.2174/092986709788681985 . ПМИД 19601803 .
- ^ Jump up to: а б «KEGG DRUG: Наркотики и психотропные вещества в Японии» . КЕГГ . 26 апреля 2024 г. Проверено 10 июля 2024 г.
- ^ Нолл Дж., Микля I (декабрь 2016 г.). «Исследование долголетия с низкими дозами селегилина/(-)-депренила и (2R)-1-(1-бензофуран-2-ил)-N-пропилпентан-2-амина (BPAP)». Науки о жизни . 167 : 32–38. дои : 10.1016/j.lfs.2016.10.023 . ПМИД 27777099 .
- ^ Фёрст С (2018). «Памяти Йозефа Нолла (1925-2018) | Венгерское общество экспериментальной и клинической фармакологии» . Проверено 10 апреля 2023 г.
- ^ Фрёстл В., Мухс А., Пфайфер А. (2014). «Когнитивные усилители (ноотропы). Часть 2: препараты, взаимодействующие с ферментами. Обновление 2014 г.». Дж. Альцгеймерс Дис . 42 (1): 1–68. дои : 10.3233/JAD-140402 . ПМИД 24903780 .
- ^ Карагеоргиу Х., Сидерис А.С., Мессари И., Лиаку К.И., Цакирис С. (август 2008 г.). «Влияние ривастигмина плюс селегилина на ацетилхолинэстеразу мозга, активность (Na, K)-, Mg-АТФазы, антиоксидантный статус и способность к обучению старых крыс» . Нервно-психические заболевания и лечение . 4 (4): 687–699. дои : 10.2147/ndt.s3272 . ПМЦ 2536534 . ПМИД 19043511 .
- ^ Столл С., Хафнер У., Пол О., Мюллер В.Е. (1994). «Возрастное снижение памяти и продолжительность жизни при лечении селегилином». Науки о жизни . 55 (25–26): 2155–2163. дои : 10.1016/0024-3205(94)00396-3 . ПМИД 7997074 .
- ^ Пуурунен К., Йолкконен Дж., Сирвиё Дж., Хаапалинна А., Сивениус Дж. (февраль 2001 г.). «Селегилин в сочетании с проживанием в обогащенной среде ослабляет дефицит пространственного обучения после очаговой ишемии головного мозга у крыс». Экспериментальная неврология . 167 (2): 348–355. doi : 10.1006/exnr.2000.7563 . ПМИД 11161623 . S2CID 22769187 .
- ^ Сабелли ХК (март 1991 г.). «Быстрое лечение депрессии комбинацией селегилин-фенилаланин» . Дж. Клин Психиатрия . 52 (3): 137. PMID 1900832 .
- ^ Сабелли Х., Финк П., Фосетт Дж., Том С. (1996). «Устойчивый антидепрессивный эффект замены ПЭА». J Нейропсихиатрия Clin Neurosci . 8 (2): 168–71. дои : 10.1176/jnp.8.2.168 . ПМИД 9081552 .
- ^ Jump up to: а б с Симпсон Х.Б., Шнайер Ф.Р., Маршалл Р.Д., Кампеас Р.Б., Вермес Д., Сильвестр Дж. и др. (1998). «Низкие дозы селегилина (L-депренил) при социофобии». Подавить тревогу . 7 (3): 126–129. doi : 10.1002/(SICI)1520-6394(1998)7:3<126::AID-DA5>3.0.CO;2-9 . ПМИД 9656093 .
- ^ Падилья СК, Виртуозо С, Тонин ФС, Борба Х.Х., Понтароло Р. (октябрь 2018 г.). «Эффективность и безопасность препаратов при синдроме дефицита внимания и гиперактивности у детей и подростков: сетевой метаанализ». Европейская детская и подростковая психиатрия . 27 (10): 1335–1345. дои : 10.1007/s00787-018-1125-0 . ПМИД 29460165 . S2CID 3402756 .
- ^ Jump up to: а б Буоли М., Серати М., Кан В. (2016). «Альтернативные фармакологические стратегии лечения СДВГ у взрослых: систематический обзор». Экспертный обзор нейротерапии . 16 (2): 131–144. дои : 10.1586/14737175.2016.1135735 . ПМИД 26693882 . S2CID 33004517 .
- ^ Блох М.Х., Панца К.Е., Ландерос-Вайзенбергер А., Лекман Дж.Ф. (сентябрь 2009 г.). «Метаанализ: лечение синдрома дефицита внимания и гиперактивности у детей с коморбидными тиковыми расстройствами» . J Am Acad Детская подростковая психиатрия . 48 (9): 884–893. дои : 10.1097/CHI.0b013e3181b26e9f . ПМЦ 3943246 . ПМИД 19625978 .
- ^ Рубинштейн С., Мэлоун М.А., Робертс В., Логан В.Дж. (август 2006 г.). «Плацебо-контролируемое исследование по изучению воздействия селегилина на детей с синдромом дефицита внимания и гиперактивности». Журнал детской и подростковой психофармакологии . 16 (4): 404–415. дои : 10.1089/cap.2006.16.404 . ПМИД 16958566 .
- ^ Ахондзаде С., Таваколян Р., Давари-Аштиани Р., Арабгол Ф., Амини Х. (август 2003 г.). «Селегилин в лечении синдрома дефицита внимания с гиперактивностью у детей: двойное слепое рандомизированное исследование». Прогресс в нейропсихофармакологии и биологической психиатрии . 27 (5): 841–845. дои : 10.1016/S0278-5846(03)00117-9 . ПМИД 12921918 . S2CID 23234928 .
- ^ Виленс Т.Э., Спенсер Т.Дж., Бидерман Дж. (март 2002 г.). «Обзор фармакотерапии взрослых с синдромом дефицита внимания и гиперактивности». Журнал расстройств внимания . 5 (4): 189–202. дои : 10.1177/108705470100500401 . ПМИД 11967475 . S2CID 37417459 .
- ^ Черемиссин О.В., Салазар Ж.О. (июнь 2008 г.). «Фармакотерапия синдрома дефицита внимания и гиперактивности у взрослых: обзор научно обоснованной практики и будущих направлений». Экспертное заключение по фармакотерапии . 9 (8): 1299–1310. дои : 10.1517/14656566.9.8.1299 . ПМИД 18473705 . S2CID 73193888 .
- ^ Jump up to: а б Мечкати Э (июль 2003 г.). «Трансдермальный пластырь с ингибитором МАО, эффективный при СДВГ» . Новости клинической психиатрии .
- ^ Jump up to: а б Хейлвуд Дж. М. (27 сентября 2018 г.). Новые подходы к фармакологическому повышению мотивации (Диссертация). Кембриджский университет. стр. 13–14. дои : 10.17863/CAM.40216 .
- ^ Jump up to: а б Каллаган К.К., Руин Дж., О'Мара С.М. (2018). «Потенциальная роль опиоидных рецепторов в мотивации и большом депрессивном расстройстве». Опиоидная система как интерфейс между когнитивной и мотивационной системами мозга . Прогресс в исследованиях мозга. Том. 239. стр. 89–119. дои : 10.1016/bs.pbr.2018.07.009 . ISBN 978-0-444-64167-0 . ПМИД 30314570 .
- ^ Йон С.Е., Рейнольдс С., Триподи Дж., Корреа М., Саламоне Дж.Д. (апрель 2018 г.). «Ингибитор моноаминоксидазы B депренил увеличивает выбор активности с высокими усилиями у крыс, протестированных с помощью процедуры выбора прогрессивного соотношения / кормления: последствия для лечения мотивационных дисфункций». Поведение мозга Res . 342 : 27–34. дои : 10.1016/j.bbr.2017.12.039 . ПМИД 29292157 .
- ^ Контрерас-Мора Х., Роуленд М.А., Йон С.Е., Корреа М., Саламоне Дж.Д. (март 2018 г.). «Частичное изменение мотивационного эффекта тетрабеназина, связанного с усилием, с помощью ингибитора МАО-В депренила (селегилина): последствия для лечения мотивационных дисфункций». Фармакол Биохим Поведение . 166 : 13–20. дои : 10.1016/j.pbb.2018.01.001 . ПМИД 29309800 .
- ^ ван Дален Дж.В., Молл ван Шаранте Э.П., Недеркурн П.Дж., ван Гул В.А., Ричард Э. (март 2013 г.). «Постинсультная апатия». Гладить . 44 (3): 851–860. дои : 10.1161/СТРОКЕАХА.112.674614 . ПМИД 23362076 .
- ^ Jump up to: а б Аль-Адави SH (1998). Нейропсихофармакология мотивации: исследование вознаграждения и лобно-подкорковых механизмов и функций (Диссертация) . Проверено 5 июля 2024 г.
- ^ Марин Р.С., Вилкош П.А. (2005). «Расстройства сниженной мотивации». J Реабилитация после травм головы . 20 (4): 377–88. дои : 10.1097/00001199-200507000-00009 . ПМИД 16030444 .
- ^ Jump up to: а б Дэн X, Шан X, Го К, Чжоу Л, Ван Ю, Ву Ю и др. (август 2023 г.). «Эффективность и безопасность антидепрессантов для прекращения курения: систематический обзор и сетевой метаанализ». Наркоман Биол . 28 (8): e13303. дои : 10.1111/adb.13303 . ПМИД 37500482 .
- ^ Jump up to: а б Хаджизаде А., Хаус С., Теодулу А., Клемперер Е., Хартманн-Бойс Дж., Ливингстон-Бэнкс Дж. и др. (май 2023 г.). «Антидепрессанты для отказа от курения» . Cochrane Database Syst Rev. 2023 (5): CD000031. дои : 10.1002/14651858.CD000031.pub6 . ПМЦ 10207863 . ПМИД 37230961 .
- ^ Кастельс Х, Касас М, Перес-Манья С, Ронсеро С, Видаль Х, Капелла Д (февраль 2010 г.). Кастельс X (ред.). «Эффективность психостимуляторов при кокаиновой зависимости». Cochrane Database Syst Rev (2): CD007380. дои : 10.1002/14651858.CD007380.pub3 . ПМИД 20166094 .
- ^ Коста А.М., Лима М.С., Мари Жде Дж. (сентябрь 2006 г.). «Систематический обзор клинического лечения сексуальной дисфункции, вызванной антипсихотиками, при шизофрении». Сан-Паулу Мед Дж . 124 (5): 291–297. дои : 10.1590/s1516-31802006000500012 . ПМИД 17262163 .
- ^ Шмидт Х.М., Хаген М., Кристон Л., Соарес-Вайзер К., Мааян Н., Бернер М.М. (ноябрь 2012 г.). «Управление сексуальной дисфункцией, вызванной терапией антипсихотиками» . Cochrane Database Syst Rev. 11 (11): CD003546. дои : 10.1002/14651858.CD003546.pub3 . ПМК 7003677 . ПМИД 23152218 .
- ^ Jump up to: а б Мерфи Б.П., Чанг Ю.К., Пак Т.В., МакГорри П.Д. (декабрь 2006 г.). «Фармакологическое лечение первичных негативных симптомов при шизофрении: систематический обзор». Шизофр Рес . 88 (1–3): 5–25. doi : 10.1016/j.schres.2006.07.002 . ПМИД 16930948 .
- ^ Jump up to: а б с Маски К., Тротти Л.М., Котагал С., Роберт Огер Р., Свик Т.Дж., Роули Дж.А. и др. (сентябрь 2021 г.). «Лечение центральных расстройств, связанных с гиперсонливостью: систематический обзор, метаанализ и оценка GRADE Американской академии медицины сна» . Джей Клин Сон Мед . 17 (9): 1895–1945. дои : 10.5664/jcsm.9326 . ПМЦ 8636345 . ПМИД 34743790 .
- ^ Jump up to: а б Нишино С., Арригони Дж., Канбаяши Т., Демент В.К., Миньо Э. (1996). «Сравнительное влияние селективных ингибиторов МАО-А и МАО-В на катаплексию собак» . Спящий Рес . 25 : 315.
- ^ Аннан Д., Мур Д.Х., Барнс П.Р., Миллер Р.Г. (июль 2006 г.). «Психостимуляторы при гиперсомнии (чрезмерной дневной сонливости) при миотонической дистрофии» . Cochrane Database Syst Rev. 2006 (3): CD003218. дои : 10.1002/14651858.CD003218.pub2 . ПМЦ 9006877 . ПМИД 16855999 .
- ^ Jump up to: а б с д Йе П.Г., Спрюйт К., ДельРоссо Л.М., Уолтерс А.С. (2023). «Повествовательный обзор менее известных лекарств для лечения синдрома беспокойных ног и патогенетических последствий их использования» . Тремор Другой Гиперкинет Мов (Нью-Йорк) . 13 :7. дои : 10,5334/том.739 . ПМЦ 9983500 . ПМИД 36873914 .
- ^ Jump up to: а б Аврора Р.Н., Кристо Д.А., Биста С.Р., Роули Дж.А., Зак Р.С., Кейси КР и др. (август 2012 г.). «Лечение синдрома беспокойных ног и расстройств периодических движений конечностей у взрослых — обновленная информация за 2012 год: параметры практики с научно обоснованным систематическим обзором и метаанализом: Руководство по клинической практике Американской академии медицины сна» . Спать . 35 (8): 1039–1062. дои : 10.5665/sleep.1988 . ПМК 3397811 . ПМИД 22851801 .
- ^ Jump up to: а б Гревал М., Хава Р., Шапиро С. (март 2002 г.). «Лечение периодических движений конечностей во сне селегилином HCl». Мов Дисорд . 17 (2): 398–401. дои : 10.1002/mds.10082 . ПМИД 11921131 .
- ^ Соарес-Вайзер К., Рэтбоун Дж., Огава Ю., Шинохара К., Бергман Х. (март 2018 г.). «Различные методы лечения поздней дискинезии, вызванной антипсихотиками» . Cochrane Database Syst Rev. 2018 (3): CD000208. дои : 10.1002/14651858.CD000208.pub2 . ПМК 6494382 . ПМИД 29552749 .
- ^ Уилкок Г.К., Биркс Дж., Уайтхед А., Эванс С.Дж. (февраль 2002 г.). «Эффект селегилина при лечении людей с болезнью Альцгеймера: метаанализ опубликованных исследований». Int J Гериатр Психиатрия . 17 (2): 175–183. дои : 10.1002/gps.545 . ПМИД 11813282 .
- ^ Сандерс О., Раджагопал Л. (июнь 2020 г.). «Ингибиторы фосфодиэстеразы при болезни Альцгеймера: систематический обзор клинических испытаний и эпидемиологии с механистическим обоснованием» . Представитель J Alzheimers Dis Rep . 4 (1): 185–215. doi : 10.3233/ADR-200191 . ПМЦ 7369141 . ПМИД 32715279 .
- ^ Лейвер К., Дайер С., Уайтхед С., Клемсон Л., Кротти М. (апрель 2016 г.). «Вмешательства по замедлению функционального снижения у людей с деменцией: систематический обзор систематических обзоров» . БМЖ Опен . 6 (4): e010767. doi : 10.1136/bmjopen-2015-010767 . ПМК 4854009 . ПМИД 27121704 .
- ^ Стинтон С., Маккейт И., Тейлор Дж. П., Лафортюн Л., Миоши Э., Мак Э. и др. (август 2015 г.). «Фармакологическое лечение деменции с тельцами Леви: систематический обзор и метаанализ» . Am J Психиатрия . 172 (8): 731–742. дои : 10.1176/appi.ajp.2015.14121582 . ПМИД 26085043 .
- ^ Беги Э., Биндер Х., Бирле С., Борнштейн Н., Дизеренс К., Гроппа С. и др. (сентябрь 2021 г.). «Руководство Европейской академии неврологии и Европейской федерации обществ нейрореабилитации по фармакологической поддержке в ранней двигательной реабилитации после острого ишемического инсульта». Эур Дж Нейрол . 28 (9): 2831–2845. дои : 10.1111/ene.14936 . ПМИД 34152062 .
- ^ Jump up to: а б Шимкович Э., Альнаггер Н., Сейфзадедарабад Ф., Кардоне П., Уайт Дж., Госсерис О. (2023). «Фармакологическое лечение». Кома и расстройства сознания (3-е изд.). Чам: Международное издательство Springer. стр. 115–146. дои : 10.1007/978-3-031-50563-8_7 . ISBN 978-3-031-50562-1 .
- ^ Jump up to: а б Масотта О, Трохано Л, Лорето В, Моретта П, Эстранео А (ноябрь 2018 г.). «Селегилин у пациентов с расстройствами сознания: открытое пилотное исследование». Может ли J Neurol Sci . 45 (6): 688–691. дои : 10.1017/cjn.2018.315 . ПМИД 30430963 .
- ^ Нолл Дж (1989). «Фармакология селегилина ((-)депренила). Новые аспекты» . Acta Neurologica Scandinavica. Дополнение . 126 : 83–91. дои : 10.1111/j.1600-0404.1989.tb01787.x . ПМИД 2515725 .
- ^ Эбади М., Шарма С., Шавали С., Эль Рефаи Х. (февраль 2002 г.). «Нейропротекторное действие селегилина». Журнал нейробиологических исследований . 67 (3): 285–289. дои : 10.1002/jnr.10148 . ПМИД 11813232 .
- ^ Jump up to: а б Бентуэ-Феррер Д., Менар Ж., Аллен Х. (1996). «Ингибиторы моноаминоксидазы B: текущий статус и будущий потенциал». Препараты ЦНС . 6 (3): 217–236. дои : 10.2165/00023210-199606030-00005 . ISSN 1172-7047 .
- ^ Нолл Дж (1978). «Возможные механизмы действия (-) депренила при болезни Паркинсона». Журнал нейронной передачи . 43 (3–4): 177–198. дои : 10.1007/BF01246955 . ПМИД 745011 .
- ^ Коэн Г., Пасик П., Коэн Б., Лейст А., Митилинеу С., Яр, доктор медицинских наук (октябрь 1984 г.). «Паргилин и депренил предотвращают нейротоксичность 1-метил-4-фенил-1,2,3,6-тетрагидропиридина (МФТП) у обезьян». Европейский журнал фармакологии . 106 (1): 209–210. дои : 10.1016/0014-2999(84)90700-3 . ПМИД 6442232 .
- ^ Финнеган К.Т., ДеЛэнни Л.Е., Ирвин И., Рикаурте Г.А., Лэнгстон Дж.В. (сентябрь 1989 г.). «Эффект истощения аминов 5,7-дигидрокситриптамина (5,7-ДГТ) у мышей C57BL/6 не увеличивается с возрастом». Исследования мозга . 496 (1–2): 251–256. дои : 10.1016/0006-8993(89)91072-x . ПМИД 2804634 .
- ^ Jump up to: а б Мораталла Р., Хайрнар А., Симола Н., Гранадо Н., Гарсиа-Монтес Х.Р., Порседду П.Ф. и др. (август 2017 г.). «Нейротоксичность амфетаминовых препаратов у человека и экспериментальных животных: основные механизмы». Прога Нейробиол . 155 : 149–170. doi : 10.1016/j.pneurobio.2015.09.011 . hdl : 10261/156486 . ПМИД 26455459 .
- ^ Jump up to: а б Пуэрта Э, Агирре Н (5 июля 2011 г.). «Метилендиоксиметамфетамин (МДМА, «экстази»): нейродегенерация против нейромодуляции» . Фармацевтика . 4 (7): 992–1018. дои : 10.3390/ph4070992 . ISSN 1424-8247 . ПМК 4058674 .
- ^ Спрэг Дж. Э., Николс Д. Е. (апрель 1995 г.). «Ингибирование МАО-Б защищает от нейротоксичности, вызванной МДМА, в полосатом теле». Психофармакология . 118 (3): 357–359. дои : 10.1007/BF02245967 . ПМИД 7542394 .
- ^ Спрэг Дж. Э., Николс Д. Е. (май 1995 г.). «Ингибитор моноаминоксидазы-B L-депренил защищает от перекисного окисления липидов, вызванного 3,4-метилендиоксиметамфетамином, и долговременного серотонинергического дефицита». J Pharmacol Exp Ther . 273 (2): 667–673. ПМИД 7538579 .
- ^ Алвес Э., Суммавиэль Т., Алвес С.Дж., Гомеш-да-Сильва Дж., Барата Х.К., Фернандес Э. и др. (сентябрь 2007 г.). «Моноаминоксидаза-B опосредует вызванное экстази нейротоксическое воздействие на митохондрии мозга подростков крыс» . Дж. Нейроски . 27 (38): 10203–10210. doi : 10.1523/JNEUROSCI.2645-07.2007 . ПМК 6672671 . ПМИД 17881526 .
- ^ Коркери Дж. М., Эллиотт С., Шифано Ф., Корацца О., Годсе А.Х. (июль 2013 г.). «MDAI (5,6-метилендиокси-2-аминоиндан; 6,7-дигидро-5H-циклопента[f][1,3]бензодиоксол-6-амин; «искра»; «минди») токсичность: краткий обзор и обновлять". Хум Психофармакол . 28 (4): 345–355. дои : 10.1002/hup.2298 . ПМИД 23881883 .
- ^ Член парламента Джонсона, Хуан XM, Николс Д.Е. (декабрь 1991 г.). «Серотониновая нейротоксичность у крыс после комбинированного лечения дофаминергическим агентом с последующим применением ненейротоксичного аналога 3,4-метилендиоксиметамфетамина (МДМА)». Фармакол Биохим Поведение . 40 (4): 915–922. дои : 10.1016/0091-3057(91)90106-c . ПМИД 1726189 .
- ^ Халладей А.К., Киршнер Э., Гессен К., Фишер Х., Вагнер Г.К. (ноябрь 2001 г.). «Роль ингибирования моноаминоксидазы и истощения запасов моноаминов в нейротоксичности, вызванной фенфлурамином, и высвобождении серотонина». Фармакол Токсикол . 89 (5): 237–248. doi : 10.1034/j.1600-0773.2001.d01-154.x (неактивен 6 июля 2024 г.). ПМИД 11881977 .
{{cite journal}}: CS1 maint: DOI неактивен по состоянию на июль 2024 г. ( ссылка ) - ^ Бенмансур С., диджей Brunswick (июль 1994 г.). «Ингибитор МАО-В депренил, но не ингибитор МАО-А клоргилин, усиливает нейротоксичность п-хлорамфетамина». Мозговой Рес . 650 (2): 305–312. дои : 10.1016/0006-8993(94)91796-5 . ПМИД 7953696 .
- ^ Спрэг Дж.Э., Джонсон, член парламента, Шмидт С.Дж., Николс Д.Е. (октябрь 1996 г.). «Исследование механизма нейротоксичности п-хлорамфетамина». Биохим Фармакол . 52 (8): 1271–1277. дои : 10.1016/0006-2952(96)00482-0 . ПМИД 8937435 .
- ^ Гольдберг С.Р., Ясар С. (1997). «Введение метамфетамина и связанная с ним нейротоксичность: эффекты селегилина (l-депренила)». Нейрохимия: клеточные, молекулярные и клинические аспекты . Бостон, Массачусетс: Springer US. стр. 327–330. дои : 10.1007/978-1-4615-5405-9_55 . ISBN 978-1-4613-7468-8 .
- ^ Ван Ф.Дж., Шиах И.С., Линь Х.К., Хуан С.Ю., Тунг К.С. (июнь 2000 г.). «Номифензин ослабляет терминальную нейротоксичность дофамина, вызванную d-амфетамином, в полосатом теле крыс». Чин Дж. Физиол . 43 (2): 69–74. ПМИД 10994696 .
- ^ Грасинг К., Азеведо Р., Каруппан С., Гош С. (январь 2001 г.). «Двухфазное воздействие селегилина на дофамин в полосатом теле: отсутствие влияния на истощение дофамина, вызванное метамфетамином». Нейрохим Рез . 26 (1): 65–74. дои : 10.1023/а:1007632700126 . ПМИД 11358284 .
- ^ Дэвидсон С., Чен К., Чжан Х., Сюн Х., Лазарус С., Ли Т.Х. и др. (ноябрь 2007 г.). «Лечение депренилом ослабляет долгосрочные пре- и постсинаптические изменения, вызванные хроническим употреблением метамфетамина». Эур Дж Фармакол . 573 (1–3): 100–110. дои : 10.1016/j.ejphar.2007.06.046 . ПМИД 17651730 .
- ^ Карвалью М., Карму Х., Коста В.М., Капела Х.П., Понтес Х., Ремиан Ф. и др. (август 2012 г.). «Токсичность амфетаминов: обновленная информация». Арка Токсикол . 86 (8): 1167–231. дои : 10.1007/s00204-012-0815-5 . ПМИД 22392347 .
- ^ Эдинофф А.Н., Суинфорд Ч.Р., Одишо А.С., Берроуз Ч.Р., Старк К.В., Раслан В.А. и др. (2022). «Клинически значимое взаимодействие лекарств с ингибиторами моноаминоксидазы» . Психологическая рес . 10 (4): 39576. doi : 10.52965/001c.39576 . ПМЦ 9680847 . ПМИД 36425231 .
- ^ Jump up to: а б «Селегилин трансдермальный (Эмсам) – Somerset Pharmaceuticals» . АдисИнсайт . 5 ноября 2023 г. . Проверено 15 июля 2024 г.
- ^ «Селегилин перорально распадающаяся таблетка (Efupi; FPF 1100 NW) – FP Pharmaceutical» . АдисИнсайт . 3 июня 2016 г. Проверено 15 июля 2024 г.
- ^ Jump up to: а б с Брюйетт Д.С., Руэль В.В., Энтрикен Т., Гриффин Д., Дарлинг Л. (март 1997 г.). «Лечение гипофизарно-зависимого гиперадренокортицизма у собак с помощью l-депренила (аниприла)». Ветеринарная клиника North Am Small Anim Pract . 27 (2): 273–286. дои : 10.1016/s0195-5616(97)50031-3 . ПМИД 9076907 .
- ^ Jump up to: а б Брэддок Дж.А., Черч Д.Б., Робертсон И.Д. (2004). «Лечение селегилином гипофизарно-зависимого гиперадренокортицизма у собак» (PDF) . Австралийский ветеринарный журнал. Архивировано из оригинала (PDF) 29 ноября 2010 года . Проверено 8 апреля 2011 г. ( PDF )
- ^ Jump up to: а б Эгианрува К (2014). Основные данные о лекарственных препаратах для рациональной терапии в ветеринарной практике . АвторДом. стр. 127–128. ISBN 978-1-4918-0010-2 .
- ^ Прпар Михевец С, Майдич Г (2019). «Когнитивная дисфункция собак и болезнь Альцгеймера – две грани одной болезни?» . Передние нейроны . 13 : 604. дои : 10.3389/fnins.2019.00604 . ПМК 6582309 . ПМИД 31249505 .
- ^ Jump up to: а б «Аниприл таблетки для животных» . Наркотики.com . Проверено 31 августа 2017 г.
- ^ Лундгрен Б. «Когнитивная дисфункция собак» . Ветеринарный партнер . Проверено 8 апреля 2011 г.
- ^ Jump up to: а б Ривьер Ж.Э., Папич М.Г. (2013). Ветеринарная фармакология и терапия . Джон Уайли и сыновья. п. 530. ИСБН 978-1-118-68590-7 .
- ^ Jump up to: а б с д Папич М.Г. (2015). Справочник Сондерса по ветеринарным препаратам: мелкие и крупные животные . Elsevier Науки о здоровье. п. 722. ИСБН 978-0-323-24485-5 .
Внешние ссылки
[ редактировать ]- Антивозрастные вещества
- Антидепрессанты
- Противопаркинсонические средства
- Афродизиаки
- Лечение синдрома дефицита внимания с гиперактивностью
- Препараты с неизвестным механизмом действия
- Энантиочистые препараты
- Ингибиторы моноаминоксидазы
- Усилители моноаминергической активности
- Нейропротекторы
- Ноотропы
- Агенты, высвобождающие норадреналин-дофамин
- Фенэтиламины
- Пролекарства
- Пропаргильные соединения
- Лиганды сигма-рецептора
- Стимуляторы
- Замещенные амфетамины
- Агонисты TAAR1
- Запрещенные вещества Всемирного антидопингового агентства